Выяснилось, как именно ацетилирование регулирует активность белка p53

Понижение активности главного антионкогена клетки белка р53 может приводить к развитию злокачественных опухолей. Ученым удалось описать новый механизм регуляции активности р53. В норме отрицательно заряженный домен белка SET связывается с положительно заряженным С-концевым доменом белка р53 и подавляет активность р53. В состоянии клеточного стресса лизиновые остатки С-концевого домена ацетилируются и комплекс с SET не образуется, а р53 остается активным. Механизм регуляции посредством ацетилирования может быть свойственен и ряду других белков.
Фактор транскрипции р53 — продукт гена ТР53, который иногда называют главным антионкогеном клетки, — экспрессируется во всех клетках организма. Механизмы функционирования этого белка изучаются много лет, но до сих пор есть много связанных с ним вопросов, на которые у ученых пока нет ответов. Известно, что если с клеткой все в порядке, то белок р53 не активен. Но при некоторых внутренних (например, при повреждении ДНК) или внешних (например, при недостатке кислорода) условиях он активируется, приобретая способность связываться с определенным участком ДНК, и может запускать транскрипцию ряда генов. В зависимости от ситуации это может приводить к замедлению процессов клеточного деления или даже к апоптозу. Подробно о функциях этого белка мы рассказывали в новости На пути к детальному каталогу раковых генов, «Элементы», 06.04.2015.
От белка p53 в буквальном смысле зависит жизнь клетки, поэтому в нормальных условиях клетки должны держать под строгим контролем его активность. А вот в условиях стрессов, которые могут привести к злокачественному перерождению клетки, этот контроль должен быть экстренно ослаблен. Ясно, что почти любое нарушение в системе контроля за активностью белка р53 может приводить к серьезным последствиям, в том числе и развитию онкологических заболеваний (см.: Главный борец с опухолями ген ТР53 может превращаться в онкоген, «Элементы», 11.11.2015): в клетках раковых опухолей часто встречаются мутации в гене ТР53.
Клетка может взаимодействовать с белками разными способами. В частности, уже синтезированные белки могут подвергаться так называемым посттрасляционным модификациям, которые способны существенно влиять на свойства белков. Один из видов таких модификаций — ацетилирование, то есть присоединение остатка уксусной кислоты. Довольно давно было показано, что ацетилирование лизинов в С-концевом домене (C-terminal domain, CTD) белка р53 приводит к тому, что у него появляется способность связываться с определенными участками ДНК (W. Gu, R. G. Roeder. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain). Делеции CTD у лабораторных мышей приводили к активации р53, что позволило предположить роль CTD как участка взаимодействия с негативными регуляторами функции р53. Но что это за регуляторы и какова роль ацетилирования CTD белка р53 во взаимодействии с регуляторами — оставалось неизвестным.
С целью получить ответы на эти вопросы группа ученых из научных учреждений США и Китая синтезировала неацетилированный (Ас−) и ацетилированный (Ас+) варианты пептида, представляющего собой CTD белка р53 (рис. 2). Эти пептиды соединили с нерастворимым носителем через биотин, позволяющий прочно фиксировать их на носителе, и проанализировали клеточные белки, способные связываться с пептидами. С Ас+ пептидом не связывались практически никакие белки, а в комплексе с CTD-Ас− обнаруживался белок молекулярного веса ~38 кДа. С помощью масс-спектрометрии пептидов, образующихся при расщеплении этого белка, он был идентифицирован как белок SET.

Рис. 2. Схема строения белка р53 и связанного с биотином С-концевого домена (CTD), использованного в экспериментах. Рисунок из обсуждаемой статьи в Nature
О белке SET известно довольно мало. Показано, что это онкопротеин, синтез которого активируется при острой миелоидной лейкемии (M. von Lindern et al. 1992. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene). И хотя уже было показано, что SET взаимодействует с белком р53 (J. Y. Kim et al. 2012. Inhibition of p53 acetylation by INHAT subunit SET/TAF-Iβ represses p53 activity), функциональные последствия этого взаимодействия и роль ацетилирования CTD белка р53 оставались неизвестными.
В обсуждаемой работе было прямо показано, что взаимодействие очищенного от лишних молекул и структур клетки белка SET с CTD белка р53 при ацетилировании последнего блокируется (рис. 1). Более того, экспрессия фермента СВР, ответственного за ацетилирование CTD белка р53, подавляла связывание SET с нормальным р53, но не с мутантным р53(в котором произошла замена лизина на аргинин), неспособным к ацетилированию. Альтернативные модификации CTD белка р53 (метилирование и ряд других) на его взаимодействие с SET не влияли. Следовательно, именно ацетилирование играет ключевую роль при взаимодействии белков р53 и SET в клетке.
SET как таковой не способен связываться с ДНК. Но в присутсвии р53 наблюдалось образование комплекса SET/р53/ДНК. Для определения влияния SET на функцию р53 как фактора активации транскрипции в клетке применили систему репортерного гена люциферазы (сделав так, что ген флюоресцирующего белка люциферазы, внедренный в клетку, оказался под контролем промотора, активируемого р53). Экспрессия люциферазы в клетке подавлялась в комбинации обычных р53 и SET, но не подавлялсь в комбинациях с лишенным CTD белком р53 или с SET, лишенным кислотного домена. В общем, полученные результаты однозначно свидетельствовали, что SET, образуя комплекс с р53, подавляет его активность, а ацетилирование CTD у р53 предотвращает образование такого комплекса, и активность р53 не подавляется.
На нескольких культурах клеток было показано, что подавление экспрессии SET с помощью siРНК, подавляющих трансляцию мРНК гена SET в белок, существенно усиливает экспрессию ряда генов, контролируемых р53. И, что соответствует функциям р53, рост клеток при этом подавлялся. В экспериментах на ксенографтных (см. Гомографт и Ксенотрансплантация) опухолях мышей при выключении SET резко подавлялся рост опухолей (рис. 3). (В данном случае в роли экспериментальных онкологических моделей — ксенографтов — выступали опухоли, вызванные пересадкой мышам клеток НСТ116, полученных из колоректальной карциномы человека.) Это подавление вызывало регрессию опухолей, в которых экспрессировался р53. У опухолей, в которых активный р53 отсутствовал, регрессии не произошло.

Рис. 3. Влияние экспрессии гена SET на рост опухолей с нормальным (слева) и подавленным (справа) геном TP53. В левый бок каждого животного были введены контрольные опухолевые клетки, в правый бок — опухолевые клетки, в которых экспрессия SET была подавлена с помощью siРНК. Внизу — выросшие опухоли. Рисунок из обсуждаемой статьи в Nature
В домене SET, взаимодействующем с р53, из 46 составляющих его аминокислот более 76% являются «кислыми». В базе данных авторы нашли еще 49 белков с подобными доменами, среди которых оказались белки, способные взаимодействовать с р53. Также как и SET, эти белки могут образовывать комплексы с неацетилированным CTD белка р53, а с ацетилированным — не могут. Авторы полагают, что регуляторная роль ацетилирования и взаимодействия белков с сильно кислыми доменами других белков не ограничивается только белком р53, а является достаточно широко распространенным механизмом регуляции (рис. 4).

Рис. 4. Общая модель зависимой от ацетилирования регуляции активности белков с лизин-обогащенными доменами (слева) путем взаимодействия с «кислыми» доменами их белков-регуляторов. Рисунок из обсуждаемой статьи в Nature
Физиологическое значение обнаруженного взаимодействия регуляторных белков с CTD белка р53 было изучено на лабораторных мышах, у которых был изменен этот домен. Авторы заменили шесть лизиновых остатков CTD белка р53 мышей на глутаминовые, сделав псевдоацетилирование лизинов, что лишило CTD способности взаимодействовать с SET. Мыши с такой мутацией погибали или внутриутробно, или через день после рождения от неконтролируемой гибели клеток мозга. Если мышата все-таки рождались, то их мозг был существенно меньше, чем у контрольных (рис. 5).

Рис. 5. a — новорожденные мыши с нормальным (p53+/+) (слева) и псевдо-ацетилированным (p53KQ/KQ) белком р53. b — мозг этих мышей. Рисунок из обсуждаемой статьи в Nature
Следовательно, в период эмбрионального развития необходим строгий контроль активности белка р53. Разрушение у мышей гена, кодирующего SET, приводило к сходным последствиям, что указывает на возможную связь наблюдавшихся эффектов именно с активностью SET. Однако пока остается недоказанным участие или неучастие в летальности каких-то других функций SET помимо взаимодействия с р53.
Высокий терапевтический противораковый потенциал ингибиторов SET авторы продемонстрировали также в экспериментах на мышах. Ранее было показано, что SET ингибирует фермент фосфатазы 2А, супрессора опухолей, подавляющего ряд сигнальных путей, которые избыточно активированны при многих раках. Следовательно, если рассмативать применение ингибиторов SET как противораковые средства, то их воздействие может не ограничиваться активацией р53. Поэтому нужно учитывать весь комплекс последствий нарушения функций SET. Кроме того, некоторые мутации р53 могут превращать этот антионкоген в онкоген (Главный борец с опухолями ген ТР53 может превращаться в онкоген, «Элементы», 11.11.2015). Разумеется, в таких случаях подавлять функцию SET нельзя. Но, в общем, применение ингибиторов SET в комбинации с активаторами ацетилирования представляется многообещающим подходом для лечения многих форм рака.
Источники:
1) D. Wang et al. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode // Nature. 2016. V. 538. P. 118–122. DOI: 10.1038/nature19759.
2) M. C. Barton. Acidic shield puts a chink in p53's armour // Nature. 2016. V. 538. P. 45–46. (Популярный синопсис к обсуждаемой статье в Nature)
Вячеслав Калинин

Последние новости

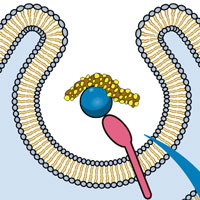


Рис. 1. Схема регуляции активности белка р53 через ацетилирование С-концевого домена. а — в условиях стресса ацетильные группы (Ас) присоединяются к шести лизиновым аминокислотным остаткам С-концевого домена белка р53 (CTD). Этот белок может связываться со специфическим участком ДНК и взаимодействовать с одним из белков-коактиваторов транскрипции — СВР или р300, которые ацетилируют гистоны. Комплекс этих событий активирует транскрипцию с промоторов, контролируемых р53. b — в отсутствие стресса С-концевой домен белка р53 не ацетилируется, и отрицательно заряженный домен белка SET, обогащенный «кислыми» аминокислотными остатками, может связываться с ним. Несмотря на то, что комплекс р53+SET по-прежнему может связываться с ДНК, он не может взаимодействовать с р300 или СВР, и поэтому транскрипция подавляется. Рисунок из синопсиса к обсуждаемой статье в Nature