Ранний рак

Задача
Онкологические заболевания (в просторечье — рак, и мы будем пользоваться этим неточным названием) — болезни, вероятность которых резко возрастает с возрастом. Например, в 20 лет раком груди не заболевает почти никто, а вероятность заболеть в 30 лет в 15–20 раз ниже, чем в 70. Однако есть небольшая группа раковых заболеваний, которые чаще возникают у детей, чем у взрослых. Например, ретинобластома (опухоль, развивающаяся из клеток сетчатки глаза) в 90–95% случаев обнаруживается в возрасте до 2-х лет.
Почему ретинобластома, в отличие от большинства видов рака, чаще встречается у детей, чем у взрослых?
Подсказка 1
Надеюсь, читатели помнят, что главная причина рака — это мутации в соматических клетках, причем обычно для превращения нормальной клетки в раковую нужно несколько мутаций.
Подсказка 2
У больных ретинобластомой опухоли обычно образуютсяодновременно в обоих глазах, причем вероятность этого тем выше, чем раньше возникла опухоль.
Подсказка 3
Есть данные, что чем больше возраст отца, тем выше риск развития ретинобластомы у его детей.
Решение
Чтобы перейти к собственно решению, нампридется вспомнить некоторые общие закономерности развития рака.
Как говорилось в подсказке, рак почти всегда связан с мутациями. Почему почти? Потому что иногда, видимо, решающую роль играет окружение клеток. Еще в 1975 году замечательная исследовательница Беатрис Минц доказала, что злокачественные клетки тератокарциномы (см. Germ cell tumor) — редкой опухоли, обычно образующейся из клеток зародышевой линии), — в составе эмбриона мыши начинают вести себя как нормальные и входят в состав нормальных органов. Более того — из потомков этих клеток могли развиваться следующие поколения здоровых мышей. Эти позднее подтвержденные работы показали, что по крайней мере некоторые клетки тератокарциномы не несут серьезных мутаций (в том числе имеют нормальное число хромосом).
Что это за мутации? Это мутации тех генов, которые придают клеткам свойства раковых: способность бесконтрольно делиться, не испытывая нужды в факторах роста и деления и не обращая внимания на соседей (то есть отсутствие контактного торможения); потенциальное клеточное бессмертие — способность делиться сколько угодно раз; неспособность «кончать жизнь самоубийством» (вступать в апоптоз), в том числе при повреждениях ДНК; как следствие этого, генетическая нестабильность — быстрое накопление всё новых и новых мутаций. Кроме того, клетки теряют признаки нормальных клеток данной ткани — становятся недифференцированными. Но всё это, как правило, еще полбеды.
Беда наступает, когда раковые клетки приобретают способность образовывать метастазы. Они выходят из собственной нормальной ткани, проникают в сосуды, разносятся по организму и поселяются в чужеродных для них тканях. А для этого им надо научиться там выживать и много чего еще уметь. Например, многие раковые клетки (не только в метастазах) стимулируют рост кровеносных сосудов, чтобы получать из них питание, и умеют подавлять иммунную атаку и даже обращать ее себе на пользу.
Не клетки, а просто тридцать три несчастья! То есть это для организма — тридцать три несчастья, а для раковых клеток-то как раз наоборот: наиболее злокачественные клетки лучше выживают, быстрее делятся и, как это ни печально, часто наиболее устойчивы к терапии.
Понятно, что столь разнообразные свойства раковых клеток зависят от разных генов и вызываются разными мутациями. Гены, непосредственно связанные с развитием рака, принято делить на две группы — онкогены и антионкогены, или гены-супрессоры опухолей.
И те, и другие выполняют в норме важные функции. Например, онкогены (точнее, их нормальные варианты — протоонкогены) воспринимают сигналы, стимулирующие выживание, рост и деление клеток, и передают их внутрь клетки. А гены-супрессоры позволяют клетке делиться только тогда, когда в ней нет серьезных повреждений ДНК. В противном случае клетка ждет, пока их удастся исправить, а если «опять ничего не получилось» — то вступает в апоптоз.
Мутации онкогенов и генов-супрессоров отличаются одной важной особенностью. Как правило, мутации онкогенов доминантные. Это означает, что мутации даже в одной копии гена (одной из родительских хромосом) достаточно, чтобы запустить раковое перерождение — в том случае, сели онкоген в ней начинает работать активнее. Такие мутации называют «мутации с избытком функции», gain-оf-function.
Их часто сравнивают с постоянно включенным газом в автомобиле (рис. 1, второй сверху). Но такой «автомобиль-клетка» не опасен для окружающих, если в нем нормально работают тормоза — гены-супрессоры. Вот если они испортились — тогда автомобиль помчится, не разбирая дороги. А мутации этих генов рецессивны. Тормоза перестанут работать, если выйдут из строя оба «контура тормозов», то есть оба аллеля — копии гена-супрессора в двух гомологичных хромосомах. Таким образом, мутации генов-супрессоров рецессивные и связаны с потерей функции (loss-of-function).
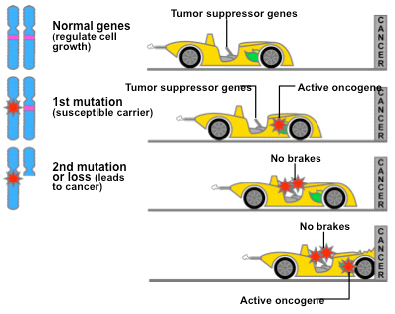
Почему же рак обычно развивается в пожилом возрасте? Одна из причин ясна. Если в клетке должно накопиться несколько мутаций, чтобы она стала раковой, то на это нужно время. Как правило, сначала в результате мутаций каких-то клеток они начинают неконтролируемо делиться, но не могут проникать в соседние ткани и кровяное русло — образуется доброкачественная опухоль (к примеру, полип в толстом кишечнике — аденома). И может пройти 10–20, а то и 30–40 лет, пока какие-то клетки этой опухоли приобретут дополнительные мутации онкогенов, генов-супрессоров и генов, связанных с образованием метастазов, — только тогда опухоль станет по-настоящему злокачественной. (Вот почему врачи непрерывно, хотя и безуспешно советуют всем людям старше 50 лет хотя бы раз в пять лет проходить колоноскопию — доброкачественные опухоли лучше вовремя обнаруживать и удалять.)
Вторая причина будет яснее, если мы прикинем вероятность возникновения мутаций, ведущих к раку. Хотя вероятность мутации конкретного гена в одной клетке очень низкая, клеток у нас слишком уж много, и они слишком часто делятся. Число клеточных делений в человеческом теле в сутки — около 2·1011. Расчеты показывают, что ежесуточно в нашем организме появляются десятки или сотни тысяч клеток с мутациями онкогенов. Многие из этих мутаций с высокой вероятностью приведут к апоптозу. А некоторые запустят постоянное деление мутантных клеток, и вскоре в каких-то из них появится вторая мутация, а потом и третья. Получается, что все мы должны были бы заболевать раком в первые годы жизни. На самом деле этого не происходит из-за контроля со стороны иммунной системы: она успевает уничтожить большинство мутантных клеток. К старости не только накапливаются мутации, но и ослабевает иммунный контроль. А если это происходит раньше — то во многих случаях (например, при СПИДе) резко повышается частота раковых заболеваний.
Теперь перейдем к рассмотрению ретинобластомы. Сразу же нужно оговориться, что рак этот — редкий, он составляет всего 3% среди всех случаев рака в первые годы жизни. В США регистрируется не более 200–300 случаев ретинобластомы в год, во всем мире — около 10 000 случаев. Это, конечно, радует, но и порождает проблемы: трудно проводить клинические испытания, а крупные компании не заинтересованы в разработке лекарств для такого редкого заболевания. Увы, многие другие виды рака встречаются в раннем возрасте гораздо чаще (на первом месте — лейкозы, на втором — опухоли мозга).
И все-таки именно на примере ретинобластомы обсуждаются и изучаются многие проблемы, связанные с раком. Во-первых, у разных пациентов болезнь протекает довольно похоже: у ретинобластомы низкая гетерогенность. Отчасти это связано с тем, что на нее слабо влияют факторы среды. Во-вторых, ее давно научились диагностировать на ранних стадиях (а значит, можно наблюдать за прогрессией опухоли). И именно для этой опухоли впервые был установлен наследственный характер и найден ген, который отвечает за ее наследование.
Тут мы уже вплотную подходим к ответу на вопрос задачи. Оказывается, около половины случаев ретинобластомы — наследственные (рис. 2, 3). Именно для ретинобластомы американский генетик Альфред Кнудсон впервые выдвинул и фактически доказал «теорию двух событий» — two-hit theory (см. Alfred G. Knudson, Jr., 1971. Mutation and Cancer: Statistical Study of Retinoblastoma — для любителей математики); см. Мутационная теория канцерогенеза. С помощью логики и достаточно простого статистического анализа он показал, что различия в сроках диагностики двусторонней и односторонней ретинобластомы, скорее всего, объясняются тем, что большинство больных двусторонней формой получили один мутантный аллель от родителя, а вторая мутация возникла в соматической клетке их сетчатки. Напротив, у многих больных с односторонней формой обе мутации возникают в соматической клетке. Позднее эта теория блестяще подтвердилась, когда был обнаружен ген ретинобластомы — RB. Это был первый открытый учеными ген-супрессор опухолей.

Рис. 2. Ретинобластома наследуется по аутосомно-доминантному типу (вверху), но сама она является рецессивным признаком (внизу). Изображение с сайтов myfamilydoctor.ru и dpuadweb.depauw.edu
Нужно отметить, что ретинобластома наследуется как аутосомно-доминантный признак, но сама она является признаком рецессивным (см. рис. 2). Это означает, что у детей человека с двусторонней ретинобластомой (которая практически всегда наследственная) вероятность получить от родителя мутантный аллель (и с вероятностью около 95% заболеть ретинобластомой) — 50% (рис. 2, вверху). Здоровый родитель, у которого мутация возникла заново в клетке эмбриона, из которой развились все его половые клетки (то есть без семейной истории заболевания), тоже с вероятностью 50% передаст мутантный аллель ребенку, и для того вероятность заболеть составит те же 95%. Но заболеет он только в тех 95% случаях, когда в одной из клеток развивающейся сетчатки мутирует второй аллель (рис. 2, внизу и рис. 3).

Рис. 3. Две мутации в гене-супрессоре необходимы для развития ретинобластомы. Чтобы черная лапа рака достала до глаза необходимы дополнительные мутации (см. Послесловие). Рисунок с сайта studyblue.com
Вероятность появления второй мутации в ходе развития глаза очень велика, поэтому наследственная форма часто (примерно в 70% случаев) билатеральная, а если опухоль возникает в одном глазу — то нередко в нескольких точках, то есть неоднократно. По крайней мере одна из опухолей обнаруживается рано, обычно на первом году жизни. Вероятность появления двух мутаций в одной соматической клетке глаза низка, и поэтому ненаследственная опухоль всегда унилатеральная (затрагивает один глаз) и унифокальная (возникает в одной точке, то есть из единственной клетки). Возникает она обычно позже и диагностируется на втором-третьем году жизни.
Такой тип развития рака объясняет и то, почему вероятность заболеть у ребенка зависит от возраста отца. Первое событие — это, как правило, точечная мутация в гене RB. А с возрастом в активно делящихся клетках — предшественниках сперматозоидов такие мутации накапливаются. Интересно, что «второе событие» при ретинобластоме часто связано не с точечной мутацией или небольшой делецией внутри гена, а с утратой гетерозиготности — делецией всего гена RB, неправильным расхождением хроматид или соматическим кроссинговером.
«Но постойте! — должен воскликнуть внимательный читатель. — Вы же ничего не объяснили! Если мутация такого важного гена у человека есть во всех соматических клетках — почему именно в глазу возникает опухоль, а не где-то еще? И почему спонтанные (ненаследственные) ретинобластомы, которых больше 50%, тоже почти всегда возникают до пятилетнего возраста, а не в старости? И почему другие формы рака не наследуются?» Попробуем объяснить, хотя бы отчасти.
Другие формы рака тоже нередко наследуются по той же схеме. В целом доля «наследственных» форм рака составляет 5–10%. И тут имеется в виду не наследственная предрасположенность, а наследование таких аллелей, при которых вероятность развития данной формы рака в течение жизни почти стопроцентная. Такие формы рака — это, например, синдром Линча (одна из разновидностей рака толстого кишечника; см. Lynch syndrome) и наследственный рак молочной железы и яичников. Они наследуются так же, как и ретинобластома (хотя возникают в более позднем возрасте). В виде исключения наследственный рак могут вызывать и наследуемые мутации онкогена, который экспрессируется в очень небольшом количестве типов клеток (например, мутации гена RET с избытком функции вызывают множественные эндокринные неоплазии). (Если зигота получает два мутантных аллеля гена RB и многих других генов-супрессоров или одну gain-of-function мутацию типичных онкогенов, зародыш гибнет в ходе эмбрионального развития. Такие мутации — эмбриональные летали.)
Мутация гена RB на самом деле влияет и на другие клетки. Например, у обладателей мутации иногда возникает «трилатеральная» ретинобластома, при которой опухоли возникают не только в глазах, но и в эпифизе (реже — в гипофизе и прилежащих участках мозга). Кроме того, частота остеосаркомы и некоторых других типов рака у обладателей этой мутации тоже резко повышена.
И все-такипочему именно ретинобластома так часто бывает наследственной и так часто возникает в раннем возрасте? Видимо, во многом это связано с особенностями сетчатки. Это небольшой по числу клеток орган, который развивается сравнительно недолго. После рождения деление клеток в сетчатке, видимо, довольно быстро останавливается. Конечно, там ищут (и находят) клетки со свойствами стволовых, которые могут делиться в пробирке; но in vivo погибшие клетки сетчатки у млекопитающих, увы, не восстанавливаются.
Раз это маленький орган с небольшим числом клеточных делений в ходе развития, то вероятность появления двух соматических мутаций в одной из его клеток мала по сравнению с другими органами. А это как раз и означает, что доля наследственных опухолей будет высокой.
Эти же факторы влияют и на частоту и время появления ненаследственных форм ретинобластомы. Давайте для наглядности сравним два органа — глаз и кишечник. В двух глазах около 500 миллионов клеток. Число клеток эпителия кишечника мне найти не удалось, но грубая прикидка показывает, что их от 10 до 100 миллиардов, то есть примерно в 100 раз больше. При этом стволовые клетки сетчатки делятся в основном во время эмбрионального развития, примерно в течение 30 недель. Клетки кишечного эпителия живут в среднем 3–4 дня, и стволовые клетки кишечника все их должны замещать. За время жизни человека они и их потомки проделывают несколько тысяч делений. Естественно, вероятность появления двух идентичных мутаций гена-супрессора в клетках кишечника высокая, и с возрастом она только растет. Во время эмбрионального развития (и вскоре после него?) мутации в двух аллелях RB в сетчатке тоже изредка возникают, но если уж до трехлетнего возраста пронесло — дальше вероятность их возникновения в неделящихся клетках почти нулевая.
Возможно, свой вклад в раннее развитие ретинобластомы вносит еще и то, что глаз — иммунопривилегированный орган. Хотя иммунная система реагирует на опухоль, но бороться с ней внутри глаза получается хуже (см., например, Kyle C. McKenna and Peter W. Chen, 2011. Influence of Immune Privilege on Ocular Tumor Development). При прочих равных в таких условиях ранние опухоли должны возникать чаще, чем в других органах. И вроде бы это правда выполняется и для других иммунопривилегированных органов — например, мозга или семенников.
Послесловие
Что же делает белок pRb — продукт гена RB? Хотя это и не имеет прямого отношения к задаче, немного в этом разберемся. Оказывается, он контролирует один из главных чекпойнтов («проверочных пунктов») клеточного цикла, а именно переход от фазы G1 к S-фазе — удвоению ДНК (рис. 4).

Рис. 4. Функция Rb — контроль перехода к S-фазе. Он подавляет активность транскрипционного фактора E2F и дополнительно меняет структуру хроматина, подавляя транскрипцию генов, необходимых для удвоения ДНК. Рисунок из статьи: F. A. Dick, 2007. Structure-function analysis of the retinoblastoma tumor suppressor protein — is the whole a sum of its parts?
Белок рRB «в норме» связывает фактор транскрипции E2F и не дает ему работать. В нормальной клетке в момент перехода к S-фазе специальные комплексы белков-циклинов и циклин-зависимых протеинкиназ (cdk) фосфорилируют pRB. В результате рRB отделяется от E2F, тот активируется и включает транскрипцию генов, отвечающих за удвоение ДНК и другие события S-фазы. Если в клетке повреждена ДНК — активируются другие белки, которые подавляют активность циклинов или cdk. То же самое происходит, когда клетка дифференцируется и кончает делиться
Дьявол сидит в деталях. И нигде эти детали не проявляются столь ярко и не оказываются такими важными, как при изучении и терапии рака. Если бы всё было так просто, как описано в решении! Хотя кому-то, может быть, этот текст и не показался слишком простым, на самом деле всё в сто раз сложнее...
Например, в тексте и на рис. 4 описаны «канонические» функции pRB. Но если открыть любой современный обзор — там описано еще с десяток других его функций. Насколько важны они для развития рака? Это вопрос, на который нет точного ответа.
Другой вопрос — есть ли третья (четвертая, пятая..., ?) мутации, которые нужны для превращения клеток ретинобластомы в раковые, или в этом исключительном случае достаточно двух мутаций в двух копиях RB? Ответ: дополнительные мутации нужны. На самом деле в результате мутации гена RB сначала возникает доброкачественная опухоль — ретинома. И только в результате следующих мутаций (но, к сожалению, очень часто) она превращается в злокачественную ретинобластому. Это могут быть мутации разных генов, и точно не известно, сколько их нужно для завершения злокачественного перерождения.
Из-за нестабильности генома клетки опухоли часто отличаются от нормальных десятками тысяч мутаций. Но как понять, сколько и какие из них — «локомотивы» рака, а сколько — «пассажиры»? Для каждой опухоли могут существовать свои комбинации критических «драйверных» мутаций. Значение может иметь и последовательность их возникновения. Только в последние годы ученые нащупывают пути к «вылавливанию» критических для развития рака комбинаций.
Возможно, для ретинобластомы нужно меньше мутаций на клетку, чем, например, для рака толстой кишки. Это может быть связано с тем, что разные гены-супрессоры работают в разных клетках (а также с условиями их работы: все-таки клеткам кишечника чаще приходится сталкиваться с разной дрянью). А генов-супрессоров в клетках тоже много, и часто они дублируют функции друг друга.
Например, и у человека, и у мыши есть три очень похожих гена — RB, он же RB1, он же pRb (ужас! надо бы все-таки генетиков заставить перейти на нормальную бинарную номенклатуру!), RBL1 (p107) и RBL2 (p130). И почему-то у человека два «гена-дублера» в сетчатке практически не работают, а у мыши — работают. Поэтому у мыши, нокаутной по гену RB (можно сделать так, чтобы он не работал только в части клеток, иначе мышь тоже погибнет до рождения), не возникают опухоли в сетчатке, но возникают в гипофизе! Для получения «мышиной» модели ретинобластомы нужно выключить хотя бы еще один ген из этой тройки — но насколько такая модель будет повторять поведение человеческой опухоли? Значит, для изучения этой опухоли надежнее использовать ортотопические ксенотрансплантаты — выращивать человеческие опухоли в глазах иммунодефицитных мышей...
Про низкую гетерогенность ретинобластомы у человека я, естественно, ничего не выдумал — это фраза из научной статьи. Но если почитать другие статьи (см., например, S. Stenfelt et al., 2017. Heterogeneity in retinoblastoma: a tale of molecules and models), то становится ясно, что тут тоже всё не так однозначно.
Во-первых, у разных людей, естественно, возникают разные дополнительные мутации при прогрессии опухоли. Кроме того, в случае ретинобластомы важную роль играют эпигенетические изменения. Например, начинается экспрессия нормального гена SYK, который обычно в сетчатке не работает; а это нарушает в клетках сетчатки запуск апоптоза.
Во-вторых, у некоторых людей с ретинобластомой не удается обнаружить мутаций в RB1. Зато у них обычно амплифицирован в большом количестве копий (до 120) онкоген MYCN (рис. 5). Белок Rb у таких людей, видимо, иногда активен — мощный мотор может пересилить работающие тормоза. Это важное открытие: такая опухоль имеет гистологические особенности, отличается ранним развитием и повышенной агрессивностью, а значит, требует более внимательного наблюдения и более агрессивной терапии.

Рис. 5. Две мутации в гене RB1 на самом деле вызывают развитие доброкачественной опухоли — ретиномы. Для ее превращения в злокачественную нужны дополнительные события — утрата или амплификация определенных участков ДНК в геноме, дополнительные мутации и эпигенетические изменения (слева). Иногда ретиноблстома развивается и у людей без мутаций в гене RB1, если в клетках их сетчатки амплифицирован онкоген MYCN (справа). Эти опухоли отличаются не только генетически, но и гистологически, и они более агрессивны. Изображение из статьи: H. Dimaras et al., 2015. Retinoblastoma
А иногда в опухоли мутации в гене RB1 не обнаруживаются, а белок Rb не активен. То ли ген не экспрессируется из-за гиперметилирования его промотора, то ли белок не активен из-за его гиперфосфорилирования. Кто виноват (какие сигнальные пути задействованы) — предстоит разбираться.
Не до конца изучен не только критически необходимый набор мутаций — точно не известно даже то, из каких клеток развивается ретинобластома. В работе 2014 года было показано, что у человека это вроде бы только дифференцирующиеся колбочки. Но эти данные были получены для клеток в культуре, а не для глаза. Для мышей ранее было показано, что это вроде бы амакриновые клетки — один из типов интернейронов глаза. Давайте найдем между колбочками человека и амакриновыми клетками мыши сходство и поймем, почему раковая опухоль развивается только из этих клеток. И такие сходства, конечно, находятся — например, повышенный уровень продукта MYCN, необходимого для выживания и деления. (Получается, что если честно отвечать на вопрос «Почему именно в глазу развивается опухоль?», то ответ будет — «Потому что некоторые клетки глаза особенные и отличаются от всех остальных».)
Но в обзоре 2017 года предполагается, что более вероятные «прародители» ретинобластомы — общие предшественники фоторецепторов и горизонтальных клеток (еще один тип интернейронов сетчатки). А в одной из работ (правда, на мышах) было сделано удивительное открытие. Оказалось, что дифференцированные горизонтальные клетки могут снова начать делиться, причем сохраняя нервные отростки и способность образовывать синапсы. И эти дифференцированные клетки образуют ретинобластому — настоящую раковую опухоль (правда, к моменту метастазирования клетки в ней становились все-таки недифференцированными). А может быть, у разных пациентов или даже у одного пациента опухоли могут возникать из разных клеток? Как это выяснить и как учитывать при лечении?
Во всём этом надо разобраться. Но пока что ученые канителятся (а они и должны канителиться — ведь ученым свойственно сомневаться, проверять и перепроверять свои данные). Медицинские же технологии не стоят на месте. В развитых странах почти 100% детей переживают свою ретинобластому, и всё большему проценту из них удается сохранить зрение. Помогают новые методы диагностики (в том числе генетическая пренатальная диагностика, позволяющая «увидеть» будущую опухоль, пока она еще невидима). Помогают новые методы облучения, новые способы доставки лекарств — внутриартериальная и внутриглазная доставка. А при экстракорпоральном оплодотворении с 2003 года доступна преимплантационная диагностика: из эмбриончиков, полученных от родителей с мутацией в RB, можно выбрать здоровые, чтобы родившийся ребенок не болел раком. Все эти методы когда-то тоже вышли из стен научных лабораторий. Так что можно не сомневаться: и с хитросплетениями генов, и с тем, как их отредактировать или отрегулировать, ученые тоже рано или поздно разберутся.
Автор благодарит Андрея Ростиславовича Зарецкого за помощь в подготовке ответа.
-
Очень интересная статья! А есть ли научные работы, в которых математически оценивается количество мутаций в клетке, необходимое для трансформации клетки в раковую (для разных типов рака)? Мне известна только одна статья: Knudson A.G., JR. Mutation and Cancer: Statistical Study of Retinoblastoma. 1971. Откуда Вами взяты цифры 3-5 мутаций на клетку?
-
Рады стараться. Цифры взяты из учебников, и имеются в виду, конечно, "драйверные" мутации - при возникновении генетической нестабильности могут накапливаться десятки, сотни и тысячи мутаций.
Математических оценок хватает, первое,что мне попалось - https://bmccancer.biomedcentral.com/articles/10.1186/1471-2407-10-3 (там есть ссылки на работы, где показано, что предположение о 5-6 мутациях при раке толстой кишки хорошо соответствуют эмпирическим данным).
-








Рис. 1. Обычно клетка становится раковой, когда в ней слишком активны онкогены и не работают гены-супрессоры опухолей. Изображение с сайта cisncancer.org