Рак против Альцгеймера: белок цистатин C активирует растворение амилоидных бляшек

Давно замечено, что у тех, кто перенес рак, почти никогда не бывает болезни Альцгеймера. Но механизм этой отрицательной связи оставался неясным. Группа китайских исследователей решила разгадать его, используя омиксные методы, позволяющие просканировать весь набор РНК и белков в клетке. Оказалось, что антиальцгеймеровский эффект раковых опухолей опосредован цистатином C — белком, который давно изучается в связи с его способностью ингибировать образование амилоида в мозге. Связываясь с амилоидом, он активирует рецептор TREM2 на мембране клеток микроглии, что заставляет их буквально поедать амилоидные бляшки, обращая патологический процесс вспять.
Практикующие врачи давно подметили интересную закономерность: люди с болезнью Альцгеймера практически никогда не имеют онкологического анамнеза. Отрицательная связь между раком и болезнью Альцгеймера была подтверждена в нескольких эпидемиологических исследованиях — например, в исследовании 2022 года, опубликованном в журнале Brain. Но не было понятно, как именно опухоли блокируют развитие болезни Альцгеймера.
Выяснить этот механизм решила группа китайских ученых под руководством Юмина Ли, невролога из Хуачжунского университета науки и технологий; их исследование недавно опубликовано в журнале Cell.
Для эксперимента, как и в предыдущих исследованиях по механизмам болезни Альцгеймера (ср. Найдены причины потери социальной памяти при болезни Альцгеймера, «Элементы», 12.12.2025), были взяты мыши с мутациями, вызывающими раннее развитие болезни Альцгеймера. Китайские исследователи использовали линию 5×FAD — мышей, несущих человеческий ген предшественника бета-амилоида человека (APP) c тремя мутациями и человеческий же ген пресенилина (PSEN1) с двумя мутациями. Шансов у таких мышей нет — у них наследственная болезнь Альцгеймера «на максималках», а мозг наполняется бляшками уже в возрасте 2-4 месяцев (для мыши это уже не детство, но довольно молодой возраст — владельцы домашних грызунов знают).
Трем опытным группам «альцгеймеровских» мышей были подсажены опухоли трех разных типов: рак легких (LLC, карцинома легких Льюиса, см. Lewis lung carcinoma), рак простаты (RM-1) и аденокарцинома толстой кишки (MC-38). Имплантация опухоли осуществлялась путем инъекции опухолевых клеток в хвостовую вену — модельные линии мышиных опухолей с током крови попадают в свою локацию, приживаются и формируют опухолевые массы, в итоге не отличаясь морфологически от «обычного» рака. Три имплантированные опухоли представляют собой карциномы, совершенно разные анатомически и клинически.
Есть у них, пожалуй, одно общее свойство: они имитируют самые распространенные и опасные типы рака у человека. И у них выявилась еще одна общая черта: все три опухоли существенно подавляли образование амилоидных бляшек в мозге альцгеймеровских мышей и замедляли развитие болезни Альцгеймера. Конечно, патология всё равно развивалась — но слабее и позже, чем у мышей из контрольной группы (рис. 1). Более того, протективный эффект сохранялся, даже если мышам не имплантировались сами опухоли, но вводились смеси секретируемых ими белков.
Стало очевидно, что какой-то из белков, секретируемых всеми тремя опухолями, ингибирует образование амилоидных бляшек. Теперь оставалось буквально просеять стог сена в поисках иголки в нём.
Сначала поиск сузили путем анализа экспрессии генов: в качестве кандидатов отбирались белки с повышенной экспрессией во всех трех типах рака. Таких набралось чуть более полутора тысяч. Затем исследователи провели протеомный анализ культуральных сред, где росли клетки всех трех типов: LLC, RM-1 и MC-38. Нашлись 94 белка, которые присутствовали во всех трех — и лишь 33 из них оказались в списке белков с повышенной экспрессией.
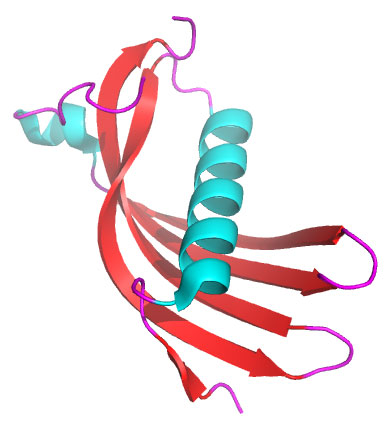
Рис. 2. Структура молекулы цистатина C. Видно, что это относительно небольшой белок — что позволяет ему проникать через почечный фильтр и через гематоэнцефалический барьер. Рисунок из Википедии
Из этого значительно более короткого списка были выбраны белки, которые ранее «засветились» в научной литературе как потенциальные регуляторы образования амилоида — их оказалось четыре. И наконец, гены этих четырех белков были по очереди «выключены» методом генного нокаута в экспериментальных мышиных опухолях, чтобы посмотреть, на каком белке исчезнет протективный эффект. Он исчез на цистатине C (Cystatin C, рис. 2) — этот белок и оказался «виновником» эффекта.
Этот белок — ингибитор сериновых протеаз, он «работает» в лизосомах и защищает клетку от переваривания собственными ферментами. Скорость его продукции в организме стабильна, и из-за небольшой молекулярной массы он проходит почечный фильтр — поэтому, наряду с креатинином, он используется в оценке функции почек (и даже является более точным маркером).
Однако уже с 2000-х годов цистатин становится известен как возможный прогностический маркер для ряда злокачественных опухолей. И в те же годы в журналах группы Nature появляются сообщения о его способности блокировать накопление бета-амилоида у «альцгеймеровских» мышей. Новизна исследования китайских ученых — в том, что они подтвердили связь онкологического и неврологического аспектов функции цистатина и показали, что именно он объясняет давно наблюдавшийся клинический эффект.
Здесь уместно вспомнить комментарий специалиста по геномике растений Сколтеха Марии Логачёвой, который она дала для заметки об эволюции роз, ранее опубликованной на «Элементах»: «Обычно все, даже лучшие статьи, публикуемые в Nature, инкрементальны. Просто в них пазл складывается наиболее красиво». В случае цистатина C пазл сложился в журнале Cell.
Однако сборкой цельной картины значимость исследования не ограничивается. Во-первых, китайские исследователи заинтересовались тем, как вообще цистатин способен оказывать влияние на бляшки — ведь он же продуцируется за пределами мозга, а мозг отделен от системного кровотока гематоэнцефалическим барьером (ГЭБ). Как оказалось в эксперименте с радиоактивно меченым цистатином, у подопытных мышей цистатин проникает через ГЭБ (рис. 3).

Рис. 3. Опухолевые клетки разных типов секретируют цистатин C, который выделяется в кровь, проникает через гематоэнцефалический барьер и способствует разрушению амилоидных бляшек в нервной ткани. Иллюстрация сделана с помощью сервиса MindTheGraph.com
Эта особенность удивила как самих исследователей, так и их коллег. Механизм этого эффекта пока неясен. Одно из возможных объяснений заключается в том, что при болезни Альцгеймера гематоэнцефалический барьер начинает функционировать хуже и становится более проницаемым. Через него, как выяснилось, проникали и другие небольшие молекулы сопоставимой молекулярной массы, например полисахарид декстран, — а вот в несколько раз больший белок альбумин уже нет. Но пока вопрос остается открытым.
Зато китайским исследователям удалось прояснить другой вопрос: как именно цистатин C подавляет развитие амилоидных бляшек. Они воспользовались методами протеомики, позволяющими получить полный набор белковых комплексов в ткани мозга подопытных мышей, и обнаружили, что в мозге цистатин C чаще всего ассоциирован с белком TREM2. Это белок-рецептор, в отношении которого у исследователей тоже давно были подозрения.
Его название расшифровывается как «триггерный рецептор 2, экспрессирующийся на миелоидных клетках» (Triggering receptor expressed on myeloid cells 2). Уже по этому названию понятно, что он является ключевым «спусковым крючком» для макрофагов нашего организма — ведь они относятся к миелоидным клеткам (то есть клеткам костного мозга, см. Myeloid tissue). В том числе — для клеток микроглии головного мозга, которые являются видоизмененными макрофагами.
Ранее уже было известно, что активация TREM2 на микроглиальных клетках способна сдерживать развитие амилоидоза мозга при болезни Альцгеймера. Работа китайских исследователей показывает, что в случае цистатина C задействуется именно этот механизм — и благодаря ему возникает значимый клинический эффект.
Дополнительные исследования показали, что цистатин C активирует TREM2 прицельно, будучи способен одновременно связываться и с TREM2, и с бета-амилоидом. Таким образом, цистатин C работает как своеобразный молекулярный «адаптер», натравливая микроглиальные клетки на амилоидные бляшки. Бляшки в таком случае буквально пожираются микроглиальными клетками (рис. 4).
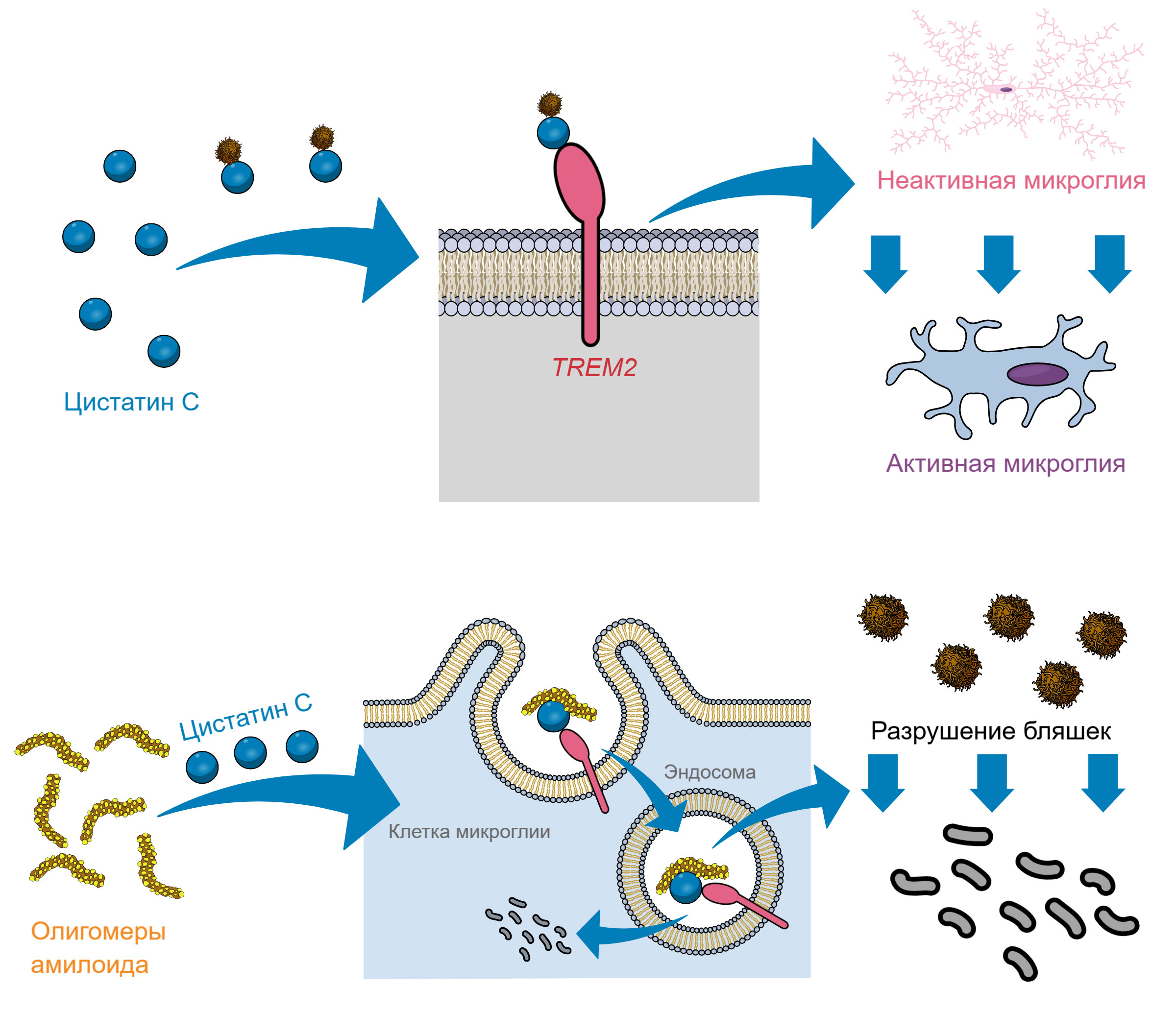
Рис. 4. Механизм антиамилоидного действия цистатина C. Вверху: цистатин C связывается с амилоидом, а затем активирует рецептор TREM2 на клетках микроглии. Это переводит микроглиальные клетки в активное состояние в непосредственной близости от бляшек. Внизу: далее микроглиальные клетки «заглатывают» небольшие фрагменты амилоида — олигомеры — связанные с цистатином C. Внутри вакуолей они разрушаются до аминокислот и безвредных коротких пептидов. Это приводит к постепенному разрушению амилоидных бляшек в мозге и не дает образоваться новым. Иллюстрация сделана с помощью сервиса MindTheGraph.com
На клеточном уровне цистатин C обращает патогенез болезни Альцгеймера вспять. Но возможно ли создать лекарство, которое было бы его аналогом и работало так же, как сам цистатин? Ответа на этот вопрос пока нет — сначала нужно найти подход к дизайну лекарства, затем проверить его в доклинических и клинических исследованиях. На этом пути возможны неприятные сюрпризы — тем более что прежние попытки разработать лекарство, активирующее TREM2, потерпели неудачу.
Дело в том, что по чисто эволюционным причинам все рецепторы в организме человека и так работают максимально эффективно даже при тяжелых заболеваниях, поэтому большинство лекарств являются блокаторами, а не активаторами ферментов и рецепторов.
Активатор сделать сложно, но теоретически можно было бы попытаться лечить болезнь Альцгеймера готовым рекомбинатным цистатином C. Но даже такой вариант потребует длительных исследований на людях: белки могут иметь капризную фармакокинетику и работать совершенно не так, как в мышиных моделях, где их продуцируют опухолевые клетки. Правда, можно использовать генную терапию, заставив клетки человека производить повышенные количества цистатина C — но такой подход технически еще сложнее и опаснее в плане развития того же рака. Кроме того, патогенез болезни Альцгеймера слишком сложен, чтобы даже полная элиминация амилоида отключила все звенья этого патогенеза.
Надежду внушает то, что эти звенья тоже исследуются, и уже открыто много альтернативных точек приложения для возможной терапии (см., например, Мутация гена фосфодиэстеразы улучшила память мышей с болезнью Альцгеймера, «Элементы», 04.04.2024), и аналоги цистатина могли бы стать дополнительной опцией в комплексном лечении.
Источник: Xinyan Li, Xiaomei Tang, Jinyu Zeng, Limin Duan, Zhenye Hou, Lanfang Li, Yiqing Guo, Changdong Chai, Jiahao Liu, Ya Wang, Ling-Qiang Zhu, Hao Li, Tongmei Zhang, Yue Wang, Aodi He, Youming Lu. Peripheral cancer attenuates amyloid pathology in Alzheimer’s disease via cystatin-c activation of TREM2. Cell, V. 189, 3, p. 853–871, 05.02.2026.
Популярный синопсис: Heidi Ledford. Cancer might protect against Alzheimer’s — this protein helps explain why. Nature.com, 22.01.2026.
Георгий Куракин
-
Для чего клеткам опухолей гиперэкспрессия цистатина? Нейросеть дает несколько вариантов, порой противоречивых с результатами этой статьи. И не все опухоли его синтезируют в должном количестве. Как-то умудрились же подобрать 3 культуры сразу в цель. И как бы дополнительный цистатин не оказался заодно антиамилоидным и проопухолевым.
-
Не думаю, что исин тут в состоянии помочь. Рано еще.
Но вопрос, конечно, интересный:
Рак случайно растворяет бляшки, или это ему необходимо уметь, чтобы существовать?
И, еще: оч прикольно б было, если вдруг окажется, что вообще эти три вида рака возникают как чрезмерная реакция организма на бляшки, как мощная его попытка их уничтожить! Некая, типа, аутоиммунная фигня)
-


А вот как бы увеличить скорость выхода результатов "в люди", в практику, в таблетки и пилюли.
Может быть ИсИн что-то тут обещает?
Уж оч хочется дожить до времен, когда врачи скажут: лечим всякий рак!
Ну, и всякий Альцгеймер - это уж мелочи...
Последние новости










Рис. 1. Микрофотографии мозга мышей, на которых амилоидные бляшки помечены двумя видами амилоид-специфичных флуоресцентных красителей и выглядят как красные и зеленые светящиеся точки. У мышей с подсаженными опухолями легких (LLC), простаты (RM-1) или кишечника (MC-38) бляшек гораздо меньше, чем у мыши из контрольной группы (Control). Иллюстрация из обсуждаемой статьи