Цифровая флуоресцентная микроскопия: новый аналитический инструмент для изучения микроорганизмов
Евгений Пучков,
Институт биохимии и физиологии микроорганизмов имени Г. К. Скрябина РАН Федерального исследовательского центра «Пущинский научный центр биологических исследований РАН» (Пущино, Россия),
Пущинский государственный естественно-научный институт (Пущино, Россия)
«Природа» №8, 2021
За последние двадцать лет флуоресцентная микроскопия трансформировалась из субъективного визуального метода в объективный аналитический подход — цифровую флуоресцентную микроскопию (ЦФМ). Произошло это благодаря совокупности инноваций, связанных с модернизацией микроскопов, использованием компьютерной обработки и анализа цифровых изображений, а также применением новых флуорофоров и методов их внедрения в клетки микроорганизмов. ЦФМ открыла принципиально новые возможности для изучения микроорганизмов, которые позволяют получать уникальную информацию о локализации и динамике внутриклеточных процессов при изучении единичных клеток микроорганизмов на субклеточном уровне.
Об авторе
 Евгений Октябринович Пучков — биофизик, доктор биологических наук, ведущий научный сотрудник, руководитель сектора консервации микроорганизмов отдела Всероссийской коллекции микроорганизмов Института биохимии и физиологии микроорганизмов имени Г. К. Скрябина РАН, профессор Пущинского государственного естественно-научного института. Область научных интересов — фундаментальные и прикладные аспекты анабиоза и гипобиоза, а также разработка и применение методов флуоресцентного зондирования и компьютерного анализа изображений в микробиологии. |
Термин «флуоресценция» ввел в 1852 г. английский ученый Дж. Стокс (George Stokes; 1819–1903), который обнаружил способность минерала флуорита светиться видимым светом при облучении невидимым ультрафиолетовым излучением. Флуоресценция — один из видов люминесценции, способности некоторых молекул после возбуждения за счет поглощения энергии в той или иной форме высвобождать часть этой энергии в виде света. Так, в некоторых химических реакциях возникает хемилюминесценция, в биологических объектах — биолюминесценция1. Есть вещества, которые испускают свет при возбуждении электрическим током (электролюминесценция), быстрыми электронами (катодолюминесценция), &γαμμα;-излучением (радиолюминесценция) и т. д. В этом контексте флуоресценция относится к категории фотолюминесценции.
Способные флуоресцировать атомы, молекулы и молекулярные комплексы называют флуорофорами или флуорохромами. Обычно этими терминами пользуются как синонимами. Однако в ряде источников под флуорохромами понимают все виды флуоресцирующих молекул, а под флуорофорами — только флуоресцирующий компонент (группировку) крупной молекулы. В микроскопии часто пользуются термином флуоресцентные красители. В классической монографии Дж. Р. Лаковица [1], посвященной различным аспектам применения флуоресценции в научных исследованиях, используется только один термин — флуорофор, для всех типов флуоресцирующих веществ. Для единообразия мы будем пользоваться этим термином. Попутно отметим, что в той же монографии можно найти сведения о физической природе флуоресценции.
Флуоресценция давно используется как источник информации в различных областях науки, в том числе в биологии и, в частности, в микробиологии. Методы изучения микроорганизмов, в которых используется феномен флуоресценции, условно можно разделить на две категории. Методы первой категории основаны на измерении таких параметров флуоресценции, как интенсивность свечения, спектры флуоресценции и возбуждения, время жизни возбужденного состояния, поляризация. По этим параметрам можно исследовать физико-химические свойства клеток или их компонентов на молекулярном уровне, используя специальные приборы — (спектро)флуориметры различной конфигурации. По техническим соображениям, связанным с чувствительностью приборов, измерения проводят на препаратах, содержащих много клеток. В итоге полученные результаты носят усредненный характер, а свойства отдельных клеток нивелируются. Методы второй категории основаны на визуализации индивидуальных флуоресцирующих клеток с использованием флуоресцентных микроскопов. С их помощью можно выявлять свечение в единичных клетках и определять пространственное распределение флуоресцирующих молекул в исследуемых объектах. Долгое время эти две категории методов существовали независимо, и в ряде случаев дополняли друг друга.
За последние двадцать лет флуоресцентная микроскопия трансформировалась из субъективного визуального метода в объективный аналитический подход — цифровую флуоресцентную микроскопию (ЦФМ). Произошло это благодаря совокупности инноваций. Во-первых, были созданы микроскопы, обеспечивающие высокочувствительную регистрацию флуоресцентных изображений в цифровом формате. Во-вторых, появились быстродействующие компьютеры, позволяющие производить в реальном времени обработку и анализ цифровых изображений. В-третьих, расширился спектр коммерчески доступных флуорофоров, а также появились новые технологии внедрения в клетки белковых флуорофоров. Всё это привело к тому, что практически все количественные аналитические методы, основанные на феномене флуоресценции [1], теперь могут быть использованы для изучения единичных клеток микроорганизмов на субклеточном уровне. В сравнении с другими имеющимися аналитическими методами исследования единичных клеток микроорганизмов [2] ЦФМ предоставляет уникальные исследовательские возможности. И не столько потому, что «увидеть — значит поверить». С помощью ЦФМ можно совмещать количественные измерения с визуальным анализом. Например, целенаправленно выбирать те или иные области клеток, в которых следует произвести измерение параметров флуоресценции. Это позволяет сопоставлять параметры флуоресценции со структурными элементами клеток.
Модернизация флуоресцентного микроскопа
Флуоресценция — довольно слабое по интенсивности излучение, особенно по сравнению с интенсивностью возбуждающего ее света. Поэтому для визуальных наблюдений в конструкции стандартного флуоресцентного микроскопа используется специальная оптическая система. Она предназначена для разделения световых потоков возбуждения и регистрации флуоресценции, отличающихся по длине волны. Совершенствование флуоресцентных микроскопов проходило по нескольким направлениям.
Прежде всего, отметим модернизацию за счет комплектации стандартных флуоресцентных микроскопов цифровыми фото- и видеокамерами. Это обеспечило регистрацию флуоресцентных изображений в цифровом формате. Такой подход позволил, с одной стороны, сделать наблюдения объективными, а также, что более важно, создал возможность обработки данных микроскопии с помощью компьютера. Важно, что с такими микроскопами можно производить также и визуальные наблюдения, в частности, для выбора объектов фото- и видеосъемки.
Другое направление связано с повышением чувствительности регистрации флуоресценции микроскопических объектов. Для этого в конструкцию флуоресцентного микроскопа были включены фотоумножители в качестве детекторов света, а также лазеры и светодиоды в качестве источников возбуждения флуоресценции. Фотоумножители способны работать в режиме подсчета единичных фотонов. Использование лазеров и мощных светодиодов вместо галогенных и ртутных ламп, которые применяются в стандартных флуоресцентных микроскопах, позволило получать для возбуждения флуоресценции интенсивное монохроматическое излучение. Фотоумножители и лазеры входят в конструкцию, например, сканирующих лазерных конфокальных микроскопов, которые имеют повышенную разрешающую способность, по сравнению со стандартными микроскопами. Кроме того, они позволяют получать изображения флуоресцирующих объектов в трехмерном пространстве.
Наконец, отметим микроскопы «особого назначения», в которых используют специальные приемы освещения и регистрации изображений. Они предназначены для уникальных конкретных задач, некоторые из которых мы рассмотрим далее. Примерами таких микроскопов могут служить микроскопы для двух(мульти)фотонной микроскопии, с полным внутренним отражением, со структурированным облучением образцов и др. Со всем «парком» современных флуоресцентных микроскопов можно познакомиться в обзоре [3].
Компьютерная обработка и анализ изображений
Математические основы обработки и анализа цифровых изображений были разработаны еще в середине прошлого века. Однако, эта методология нашла широкое применение во многих областях науки и практики только с появлением доступного компьютерного оборудования необходимой мощности. Общие принципы компьютерной обработки и анализа цифровых изображений в приложении к микроскопии достаточно подробно изложены в [4–6]. Кратко их можно представить следующим образом [7].
Изображения в цифровом формате представляют собой набор элементов, называемых пикселями, если изображение получено в двумерном (2D) пространстве, или вокселями, если изображение получено в трехмерном (3D) пространстве. Каждый пиксель (воксель) содержит закодированную в цифровом виде информацию о его X-, Y- и Z-положении в декартовых координатах, а также об оптических свойствах в этой точке пространства. Характер оптической информации зависит от того, какая оптическая система была использована при регистрации изображения. Если эта информация была получена с помощью монохромных цифровых фото- и видеокамер или фотоумножителя, то она представляет собой данные об интенсивности света в единицах шкалы серого цвета. Если изображения были получены с использованием цветных фото- и видеокамер, тогда они представляют собой матрицу пикселей с информацией, закодированной по одной из моделей «цветовых пространств» — RGB, HSV, CIE-Lab или YCrCb. RGB (аббревиатура английских слов red, green, blue — красный, зеленый, синий) — одна из наиболее популярных моделей, в которой используется 24-битная комбинация интенсивностей красного, зеленого и синего цветов с 8 битами на каждый цвет. Исходные данные в цифровом формате обрабатываются компьютером как параметры специальной математической модели, сконструированной из различных алгоритмов. Обработка исходных данных с помощью этой модели позволяет ими манипулировать таким образом, чтобы улучшить визуальное восприятие всего изображения или отдельных компонентов (рис. 1). Кроме того данные, закодированные в цифровом изображении, можно использовать для их количественного анализа. Этот подход называют компьютерным анализом изображений. С помощью компьютерного анализа изображений определяют оптические свойства объектов в сопряжении с их пространственными характеристиками.
Флуорофоры
Естественных флуорофоров в живых клетках сравнительно немного. Кроме того, спектральные свойства их свечения таковы, что ими практически невозможно воспользоваться для микроскопии. Поэтому для флуоресцентной микроскопии используют специально синтезированные флуорофоры. Низкомолекулярные синтетические флуорофоры условно делят на две категории — зонды и метки. Флуоресцентные зонды способны с некоторой степенью специфичности вступать в нековалентное взаимодействие с определенными компонентами клеток, при этом параметры их флуоресценции меняются. Например, существуют зонды для исследования свойств мембран, нуклеиновых кислот, некоторых внутриклеточных органелл, pH цитоплазмы, содержания в ней некоторых неорганических ионов и др. Флуоресцентные метки способны специфически ковалентно связываться с определенными макромолекулами (белками, липидами, углеводами), придавая им флуоресцентные свойства. В настоящее время имеется богатый выбор коммерчески доступных флуоресцентных зондов и меток [8].
Использование многих флуоресцентных зондов или меченых макромолекул затруднено или даже невозможно при исследовании интактных (нефиксированных) клеток. Это связано с избирательной проницаемостью цитоплазматической мембраны, через которую в клетку не могут попасть многие гидрофильные флорофоры и уж тем более меченые макромолекулы. Одно из возможных решений — создание гидрофобных производных гидрофильных флорофоров, у которых после проникновения в клетки гидрофобная надстройка отщепляется внутриклеточными ферментами (например, эстеразами) [8]. Особую роль в решении этой и многих других проблем внутриклеточного флуоресцентного зондирования сыграли две технологии, основанные на применении флуоресцентных белков (ФБ).
Первая такая технология была создана благодаря обнаружению и клонированию природного белка медузы Aequorea victoria, флуоресцирующего в зеленой области спектра, известного как зеленый флуоресцентный белок (ЗФБ; англ. Green Fluorescence Protein, GFP) [9]. Было установлено, что клонированный ген ЗФБ является самодостаточным для биосинтеза полноценного флуоресцирующего белка в различных клетках. Это послужило предпосылкой для разработки молекулярно-биологических методов, с помощью которых ЗФБ можно присоединять к другим белкам (англ. protein fusion — слияние белков) в качестве флуоресцентной метки. Позднее были обнаружены природные и синтезированы искусственные гомологи ЗФБ, способные флуоресцировать в более длинноволновой области спектра (рис. 2). Их тоже можно использовать для создания гибридных ФБ. Подробнее с историей разработок и принципами технологии создания ФБ на основе гомологов ЗФБ можно познакомиться в обзорах [12, 13].

Рис. 2. Модель структуры зеленого флуоресцентного белка (а; в центре полипептидного «бочонка» расположен флуорофор [10]) и палитра генноинженерных флуоресцентных белков (б; слева направо: производные GFP — EBFP, ECFP, EGFP, YFP; производные mRFP1 — mHoneydew, mBanana, mOrange, tdTomato, Tangerine, mStrawberry, mCherry mGrape1, mRaspberry, mGrape2, mPlumm; [11] с изменениями)
Принцип второй технологии основан на создании гибрида внутриклеточного целевого белка со специальной белковой надстройкой (англ. tag — метка, маркер), обладающей способностью ковалентно присоединять к себе низкомолекулярные флуорофоры. Благодаря этой способности «метить себя» в англоязычной литературе такие белки называют self-labeling proteins. Они представляют собой ферменты, которые ковалентно присоединяют к себе флуоресцентные фрагменты специально для этого модифицированных субстратов. Технология включает два этапа. Сначала методами молекулярной биологии в исследуемых клетках создают гибрид целевого белка с соответствующей надстройкой. Затем добавляют субстрат, который содержит необходимый флуорофор. В результате вся конструкция, включающая целевой белок, оказывается меченной экзогенным флуорофором.
Один из популярных белков, способных «метить себя» флуорофорами, разработан на основе O6-алкил гуанин-ДНК алкилтрансферазы человека. Установлено, что присоединение этого фермента к целевым белкам в клетках бактерий, дрожжей и линии CHO2 и введение флуоресцентных производных субстрата этого фермента O6-бензил гуанина приводит к флуоресцентному мечению гибридных белков [14]. Вслед за этими исследованиями был получен генноинженерный продукт гена O6-алкил гуанин-ДНК алклилтрансферазы, SNAP-tag, который в настоящее время коммерчески доступен и входит в состав различных экспрессионных векторов. Его субстратом служат флуоресцентные производные O6-бензил гуанина (рис. 3) [15]. Имеются и другие системы мечения внутриклеточных белков экзогенными флуорофорами с помощью этой технологии, например, CLIP-tag и Halo-tag. Их субстратами становятся флуоресцентные производные O6-бензил цитозина [16] и первичных алкилгалидов [17], соответственно.

Рис. 3. Ковалентное мечение целевого белка путем присоединения надстройки SNAP-tag, генноинженерного продукта гена O6-алкил гуанин-ДНК алклилтрансферазы, с последующей добавкой флуоресцентного производного О6-бензил гуанина ([15] с изменениями). В качестве флуорофора может быть, например, флуоресцеин
Технология создания ФБ с помощью белковых надстроек, способных «метить себя» флуорофорами, имеет несколько преимуществ по сравнению с технологией на основе гомологов ЗФБ. Полученные таким образом ФБ обладают большим квантовым выходом и повышенной стабильностью при возбуждении высокоинтенсивными источниками света [18]. Кроме того, возможность использования по этой технологии обширного набора коммерчески доступных низкомолекулярных флуорофоров [8] открывает новые направления исследований.
Вместе с тем следует отметить и наличие проблем при использовании этой технологии на микроорганизмах. Прежде всего, это трудности ее применения для исследования живых (нефиксированных) клеток. Они связаны с непроницаемостью мембран и/или клеточных стенок бактерий и дрожжей для большинства гидрофильных флуоресцентных субстратов. Обойти эту трудность позволяет, например, использование гидрофобных производных флуорофоров или электропорации. Следует помнить и о том, что обе технологии создания ФБ подразумевают модификацию целевых белков, что необходимо учитывать при интерпретации полученных с их помощью данных.
Интересные экспериментальные возможности появились после обнаружения и синтеза фоторегулируемых (англ. photoswitchable) ФБ. Существуют белки, которые исходно не флуоресцируют, но с помощью фотоактивации могут приобрести такую способность — необратимо (например, фотоактивируемый ЗФБ) или обратимо (например, белок Dronpa). Другие ФБ при соответствующем облучении могут необратимо менять спектральные свойства. Например, ФБ EosFP, флуоресцирующий зеленым светом, после облучения синим светом начинает флуоресцировать красным (рис. 4). Свойства различных фоторегулируемых ФБ рассмотрены в обзорах [19, 20].
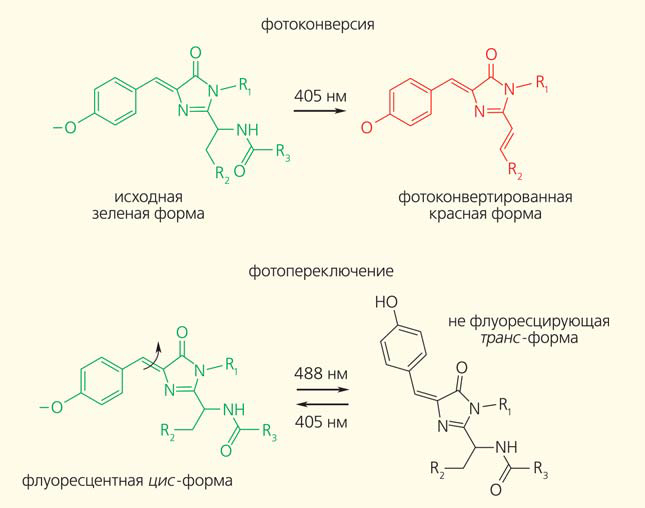
Рис. 4. Примеры фоторегуляции некоторых ФБ (показаны только флуорофоры; [19] с изменениями): а — ФБ EosFP способен необратимо конвертироваться из зеленой флуоресцентной формы в красную флуоресцентную форму при облучении синим или ультрафиолетовым светом; б — ФБ Dronpa при облучении определенными длинами волн может подвергаться обратимой цис-транс-изомеризации. При этом только цис-форма обладает способностью флуоресцировать
Одним из важнейших результатов применения технологий формирования внутриклеточных ФБ и фоторегулируемых флуорофоров стало создание методов оптической микроскопии сверхвысокого разрешения (ОМСР).
Оптическая микроскопия сверхвысокого разрешения
Разрешающая способность стандартной оптической микроскопии ограничена дифракционными свойствами света. Этот так называемый дифракционный предел пространственного разрешения приблизительно равен половине длины световой волны используемого освещения. На практике это означает, что с помощью стандартного оптического микроскопа невозможно различить два объекта, которые расположены ближе, чем половина длины волны используемого света (в большинстве случаев это несколько сотен нанометров) [3]. Методы оптической микроскопии сверхвысокого разрешения позволяют преодолеть этот предел различными способами. Впервые такая возможность была продемонстрирована с помощью так называемой ближнепольной микроскопии (англ. near-field microscopy). Она основана на регистрации световых потоков специальным оптическим зондом, который находится на расстоянии от поверхности образца меньше длины волны используемого света. Однако более практичными для изучения биологических объектов оказались методы, основанные на обычной дальнопольной микроскопии (англ. far-field microscopy). Для их реализации не требуются микроскопы со специальным оптическим зондом, и они обеспечивают возможность исследовать не только поверхностные, но и более глубокие области препарата. Чтобы подчеркнуть возможность исследования объектов за пределами «дифракционного барьера» с разрешением в нанометровом диапазоне был введен термин «дальнопольная оптическая наноскопия» (англ. far-field optical nanoscopy) [21].
Условно методы ОМСР можно разделить на две категории.
— Методы, основанные на локализации единичных молекул: микроскопия локализации путем фотоактивации флуоресценции (англ. fluorescence photoactivation localisation microscopy, fPALM/PALM) [22, 23] и микроскопия стохастической оптической реконструкции (англ. stochastic optical reconstruction microscopy, STORM) [24, 25]. Общий принцип, который лежит в основе этой категории методов, заключается в следующем. Флуоресцирующие молекулы в исследуемом образце возбуждаются таким образом, чтобы одновременно могла флуоресцировать только часть из них. Эта процедура проводится многократно с регистрацией изображений. Полученные изображения накладываются друг на друга. Результирующее изображение включает все флуоресцирующие молекулы, в том числе и те, которые расположены на таком близком расстоянии, что при обычной микроскопии они были бы неразличимы (рис. 5). Для реализации этого принципа в методах fPALM/PALM используются специальные фоторегулируемые ФБ, которые могут «включаться/выключаться» определенными комбинациями возбуждающего излучения [26]. В методах STORM применяют специально подобранные пары флуорофоров, один из которых выступает в роли «активатора», а другой «акцептора» возбуждения [24, 25]. Разрешающая способность PALM/STORM наноскопии составляет около 20–60 нм.

Рис. 5. Схематическая иллюстрация методов, основанных на локализации единичных молекул, с использованием микроскопов, оборудованных системой облучения в режиме полного внутреннего отражения ([20] с изменениями). Исследование проводится путем осуществления нескольких циклов фотоактивации небольшой фракции флуорофоров во фронте затухающей волны, возбуждения их флуоресценции, регистрации их локализации по изображениям и тушения (обесцвечивания). Затем проводится компьютерная реконструкция путем наложения многих сотен или даже тысяч изображений друг на друга. В результате можно выявить и различить флуорофоры, расположенные на расстоянии меньше дифракционного предела разрешения, поскольку они флуоресцировали на разных циклах фотоактивации, возбуждения и обесцвечивания
— Методы, основанные на специальном освещении образцов, такие как микроскопия со стимулируемым подавлением излучения (англ. stimulated emission depletion microscopy, STED) [21, 27] и микроскопия со структурированным освещением (англ. structured illumination microscopy, SIM) [28]. Для реализации этой категории методов не требуется сверхвысокая чувствительность регистрации свечения отдельных молекул. При микроскопии STED используют облучение двумя импульсами лазера. Первый — возбуждает флуорофоры до флуоресцирующего состояния, второй — применяется для подавления флуоресценции у части флоурофоров в области, окружающей узкую область фокусировки объектива. В таком режиме освещения проводится сканирование образца и формируется флуоресцентное изображение с разрешением 30–60 нм (рис. 6). В микроскопах SIM поток возбуждающего света проходит через специальную оптическую решетку. Это приводит к формированию в фокальной плоскости образца пространственно структурированного освещения за счет интерференционного (муарового) эффекта. После обработки специальными компьютерными программами серии полученных таким образом флуоресцентных изображений образца можно улучшить разрешение приблизительно до 100 нм.
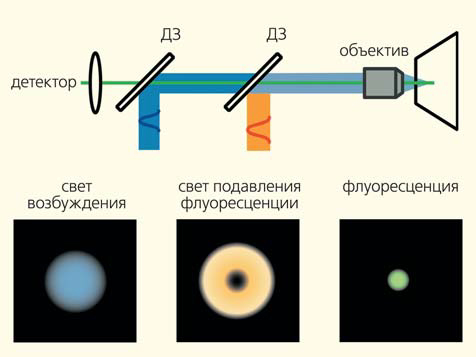
Рис. 6. Оптическая схема микроскопии со стимулируемым подавлением флуоресценции (а) и профили световых потоков (б) [20]. ДЗ — дихроическое зеркало. Импульсы света возбуждения и подавления флуоресценции (синий и оранжевый цвета, соответственно) с пикосекундным интервалом подаются на исследуемый объект. Свет флуоресценции (зеленая линия) регистрируется фотодетектором (фотоумножителем)
Проиллюстрируем примерами возможности разнообразных методов ЦФМ для изучения микроорганизмов на субклеточном уровне.
Изучение дрожжей и бактерий на субклеточном уровне

Рис. 7. Определение вязкости в вакуолях дрожжей Saccharomyces cerevisiae c помощью ЦФМ по броуновскому движению комплексов неорганических полифосфатов — КНПФ (подробнее см. [30]): а — флуоресцентная микрофотография клетки S. cerevisiae после окраски ДАФИ (4’,6-диамидино-2-фенилиндол); комплекс полифосфатов флуоресцирует желтым светом (показан стрелкой), ядра и митохондрии флуоресцируют синим светом; б — размер КНПФ определялся по профилю флуоресценции; в — положения КНПФ фиксировались на последовательных кадрах скоростной фотосъемки с интервалом 0.43 с, и по ним определялось смещение; г — измерения размеров и среднего квадрата смещения КНПФ с помощью компьютерного анализа изображений позволили рассчитать вязкость внутри вакуолей четырех клеток (показаны стрелками)
Дрожжи. Показана возможность проведения субклеточной микрофлуориметрии единичных клеток дрожжей Saccharomyces cerevisiae c помощью ЦФМ. Были выполнены измерения вязкости в вакуолях по броуновскому движению комплексов неорганических полифосфатов (рис. 7) и поляризации флуоресценции хинакрина (рис. 8). Другие методы измерения внутриклеточной вязкости описаны в обзоре [29]. Кроме того, был разработан подход, названный псевдоспектральным анализом, который позволил локализовать внутриклеточное распределение противоракового соединения доксорубицина в клетках дрожжей, как модели клеток животных и человека [30].
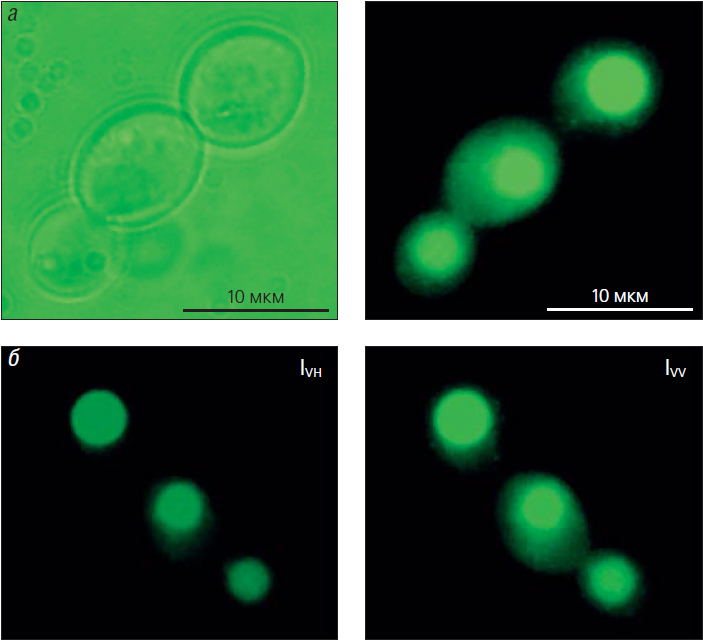
Рис. 8. Определение вязкости в вакуолях дрожжей Saccharomyces cerevisiae c помощью ЦФМ по анизотропии флуоресценции хинакрина (подробнее см. [30]): а — микрофотографии клеток S. cerevisiae, обработанных хинакрином, в режиме светопропускания (слева) и флуоресценции (справа); хинакрин, накопленный в вакуолях, флуоресцирует ярким желто-зеленым светом; б — флуоресцентные микрофотографии клеток S. cerevisiae с хинакрином в вакуолях, зарегистрированные на микроскопе с поляризаторами в каналах возбуждения и регистрации флуоресценции; слева — плоскости поляризаторов в перпендикулярном положении, справа — плоскости поляризаторов в параллельном положении. Измерения анизоторопии флуоресценции c помощью компьютерного анализа изображений позволили рассчитать вязкость внутри вакуолей 39 клеток
ЦФМ позволила по-новому подойти к проведению и использованию морфометрии дрожжевых клеток. Созданы специальные компьютерные программы для количественной обработки данных по внутриклеточной локализации ядер, актина, митохондрий, вакуолей, эндоплазматического ретикулума, аппарата Гольджи, эндосом, веретина и септина, меченных различными флуорохромами и ФБ в совокупности с морфометрическими показателями геометрии клеток [31, 32]. Эти программы стали основой базы морфологических данных гаплоидных клеток S. cerevisiae с нелетальными мутациями для изучения взаимосвязи функций определенных генов с морфологическими признаками [33]. Комплексная морфометрия на основе ЦФМ может найти применение в прикладных исследованиях. Например, показана возможность использования морфометрических показателей дрожжевой формы грибов и методов искусственного интеллекта для разработки противогрибковых препаратов [34].
Имеются примеры использования ОМСР в исследованиях дрожжевых клеток. Конфокальная микроскопия и ОМСР применялись для изучения локализации макромолекул, а также динамики внутриклеточных процессов на дрожжах S. pombe. Были проведены измерения расстояний между кластером генов Chr1, цитоплазматической мембраной и полярным тельцем веретена в зависимости от условий культивирования клеток [35]. fPALM использовали для изучения структуры и динамики сборки/распада интерфазных узелков3 [36], механизма полимеризации нитей актина при эндоцитозе, индуцированном клатрином [37]. С помощью SIM удалось выявить пространственное взаимодействие белковых компонентов при сборке центросом в ходе деления [38].
Объемные (3D) изображения живых (нефиксированных) клеток S. cerevisia, обеспечивающих чувствительность единичных молекул, можно получить методом, который основан на применении специального микроскопа. В его канале регистрации изображения создается астигматизм, что обеспечивает возможность получения 3D координат единичных флуорофоров. С помощью этого метода исследовалась динамика связывания белка Mig1 (Zn-содержащий фактор транскрипции), меченного ЗФБ, с ДНК клеток. Полученные данные использовали для характеристики 3D распределения мест связывания белка в ДНК, а по ним была предсказана 3D архитектура генома [39].
Бактерии. Одно из первых исследований бактерий на субклеточном уровне с помощью ЦФМ было связано с обнаружением трансформации зеленого флуоресцентного белка в красный флуоресцентный белок при фотоактивации светом в области 475–495 нм (рис. 9,а). Это свойство было использовано для измерения скорости внутриклеточной диффузии белка в клетках Escherichia coli (рис. 9,б) [40].
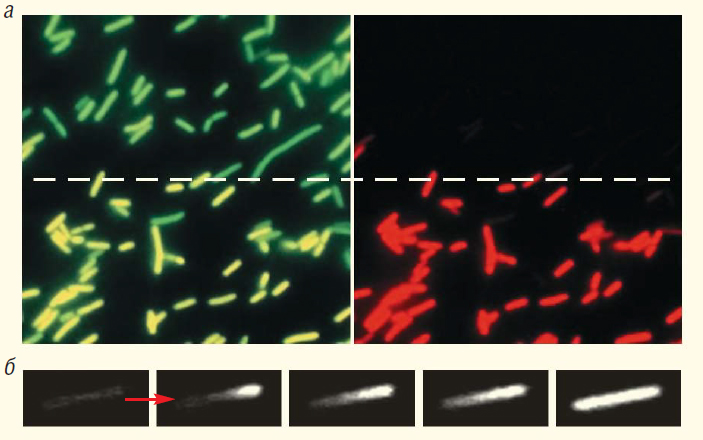
ОМСР с помощью ЦФМ обеспечила уникальную возможность регистрации динамики отдельных молекул в единичных клетках бактерий, размеры которых находятся на пределе разрешающей способности оптической микроскопии. К примеру, с помощью fPALM были исследованы процессы репарации ДНК на живых (нефиксированных) клетках E. coli по диффузионным характеристикам единичных молекул ДНК полимеразы и лигазы [41]. Для этого использовали мечение целевых белков флуоресцентными белками. Так, в частности, измеряли траектории ДНК полимеразы Pol1, меченной флуоресцентным белком PAmCherry, до и после повреждений ДНК, вызванных метилметансульфонатом. Оценка коэффициентов диффузии индивидуальных треков Pol1 в 2D пространстве показала, что при наличии повреждений в ДНК активируется взаимодействие Pol с ДНК. Об этом свидетельствовало увеличение количества участков замедленной диффузии Pol1 (рис. 10).

Рис. 10. Взаимодействие ДНК полимеразы Pol1, меченной флуоресцентным белком PAmCherry, с ДНК при репарации повреждений, вызванных метилметансульфонатом (MMС) в клетках E. coli, по данным fPALM ([41] с изменениями): а — построение карты траекторий единичных молекул Pol1 по данным fPALM при повреждении ДНК, вызванной ММС; б — карта траекторий единичных молекул Pol1 в клетке; в — количественная оценка репарационной активности Pol1 после обработки клеток ММС по коэффициенту диффузии. Синим цветом обозначены движения свободных молекул Pol1, красным — молекул Pol1, связанных с ДНК
Особый интерес представляет возможность одновременной регистрации взаиморасположения структурных компонентов бактериальных клеток с разрешением в нанометровом диапазоне. С этой целью была разработана методика получения флуоресцентных изображений нескольких отдельных молекул с одновременным флуорохромированием структурных элементов клеток [42]. Методика основана на fPALM с использованием фоторегулируемых белков, которыми метят целевые белки. При этом используется комбинация фотоконверсии и фотоактивации путем варьирования режимов облучения различными длинами волн. Это дает возможность регистрировать даже близкорасположенные флуорофоры в одном оптическом канале регистрации (рис. 11). Было показано также, что этот подход может быть использован и на других типах клеток, в частности, на дрожжах.

Рамки статьи не позволяют привести все примеры широкого спектра возможностей ЦФМ для изучения бактерий. Отметим только, что применение различных методов ЦФМ в исследованиях бактерий на субклеточном уровне отражено в ряде обзоров, посвященных, в частности, изучению структуры цитоскелета и нуклеоида [43]; динамики транскрипции [44] и трансляции [45]; динамики взаимодействия белков с ДНК [46]; репликации и репарации ДНК [47], организации систем секреции и внутриклеточной компартментализации [48]; внутриклеточной сигнализации и динамики экспрессии генов [49]; механизмов действия антимикробных соединений [50]; а также техническим аспектам [51] и перспективам использования ЦФМ в изучении внутриклеточной биохимии на уровне единичных молекул [52].
* * *
ЦФМ открыла две принципиально новых возможности для изучения микроорганизмов. Первая — это измерение параметров флуоресценции целевых флуоресцентных зондов и меток, связанных с определенными структурами единичных клеток. Вторая — расширение границ визуализации внутриклеточных компонентов за пределы «дифракционного барьера» световой микроскопии в нанометровый диапазон. Это позволяет получать уникальную информацию о локализации и динамике внутриклеточных процессов на молекулярном уровне.
К настоящему времени разработано много методов, основанных на ЦФМ, для решения широкого спектра задач, связанных со структурно-функциональной организацией микробных клеток. Вместе с тем идет непрерывное развитие этой методологии за счет разработки новых программных средств, совершенствования микроскопов, а также методов флуоресцентного зондирования целевых компонентов клеток.
ЦФМ позволяет совмещать количественные измерения с визуальным анализом, а также сопоставлять параметры флуоресценции со структурными элементами клеток. Важно также то, что многие методики ЦФМ могут быть реализованы на живых (нефиксированных) клетках. Благодаря этому открывается перспектива исследования динамики метаболических процессов на субклеточном уровне. Всё это определяет особое место ЦФМ в современном наборе аналитических инструментов изучения единичных клеток микроорганизмов [2].
Литература / References
1. Lakowicz J.R. Principles of Fluorescence Spectroscopy. Berlin, 2006.
2. Puchkov E. Analytical Techniques for Single-Cell Studies in Microbiology. Handbook of Single Cell Technologies. T. Santra, F. G. Tseng (eds.). Singapore, 2021; 1–32. DOI:10.1007/978-981-10-4857-9_17-3.
3. Sanderson M.J., Smith I., Parker I. et al. Fluorescence microscopy. Cold Spring Harb Protoc. 2014; 10: pdb.top071795. DOI:10.1101/pdb.top071795.
4. Sbalzarini I.F. Seeing is believing, quantifying is convincing, computational image analysis in biology. Adv. Anat. Embryol. Cell Biol. 2016; 219: 1–39. DOI:10.1007/978-3-319-28549-8_1.
5. Nketia T.A., Sailem H., Rohde G. et al. Analysis of live cell images. Methods, tools and opportunities. Method. 2017; 115: 65–79. DOI:10.1016/j.ymeth.2017.02.007.
6. Wallace C.T., Jessup M., Bernas T. et al. Basics of digital microscopy. Curr. Protoc. Cytom. 2018; 83: 12.2.1–12.2.14. DOI:10.1002/cpcy.31.
7. Puchkov E. Quantitative Optical Microscopy in Microbiology. An Introduction to Microorganisms. Q-S. Wu, Y-N. Zou, F. Zhang, B. Shu (eds). N.Y., 2021; Chapter 1: 1–31.
8. The Molecular Probes Handbook. A Guide to Fluorescent Probes and Labeling Technologies. I. Johnson, M. Spence (eds.). Moscow: Life Technologies, 2010.
9. Tsien R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998; 67: 509–544. DOI:10.1146/annurev.biochem.67.1.509.
10. Remington S.J. Green fluorescent protein: a perspective. Protein Sci. 2011; 20(9): 1509–1519. DOI:10.1002/pro.684.
11. Tsien R.Y. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005; 579(4): 927–932. DOI:10.1016/j.febslet.2004.11.025.
12. Snapp E. Design and use of fluorescent fusion proteins in cell biology. Curr. Protoc. Cell Biol. 2005; Chapter 21: 21.4.1–21.4.13. DOI:10.1002/0471143030.cb2104s27.
13. Campbell R.E. Fluorescent proteins. Scholarpedia. 2008; 3(7): 5410. DOI:10.4249/scholarpedia.5410.
14. Keppler A., Gendreizig S., Gronemeyer T. et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003; 21: 86–89. DOI:10.1038/nbt765.
15. Juillerat A., Gronemeyer T., Keppler A. et al. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003; 10(4): 313–317. DOI:10.1016/S1074-5521(03)00068-1.
16. Gautier A., Juillerat A., Heinis C. et al. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008; 15: 128–136. DOI:10.1016/j.chembiol.2008.01.007.
17. Los G.V., Encell L.P., McDougall M.G. et al. HaloTag, a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008; 3(6): 373–382. DOI:10.1021/cb800025k.
18. Hinner M.J., Johnsson K. How to obtain labeled proteins and what to do with them. Curr. Opin. Biotechnol. 2010; 21(6): 766–776. DOI:10.1016/j.copbio.2010.09.011.
19. Chozinski T.J., Gagnon L.A., Vaughan J.C. Twinkle, twinkle little star, photoswitchable fluorophores for super-resolution imaging. FEBS Lett. 2014; 588(19): 3603–3612. DOI:10.1016/j.febslet.2014.06.043.
20. Minoshima M., Kikuchi K. Photostable and photoswitching fluorescent dyes for super-resolution imaging. J. Biol. Inorg. Chem. 2017; 22(5): 639–652. DOI:10.1007/s00775-016-1435-y.
21. Hell S. Far-Field Optical nanoscopy. Science. 2007; 316(5828): 1153–1158. DOI:10.1126/science.1137395.
22. Betzig E., Patterson G.H., Sougrat R. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006; 313(5793): 1642–1645. DOI:10.1126/science.1127344.
23. Hess S.T., Girirajan T.P., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006; 91(11): 4258–4272. DOI:10.1529/biophysj.106.091116.
24. Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods. 2006; 3(10): 793–795. DOI:10.1038/nmeth929.
25. Bates M., Huang B., Dempsey G.T. et al. Multicolor super-resolution imaging with photoswitchable fluorescent probes. Science. 2007; 317(5845): 1749–1753. DOI:10.1126/science.1146598.
26. Heilemann M., Dedecker P., Hofkens J. et al. Photoswitches: key molecules for subdiffraction resolution fluorescence imaging and molecular quantification. Laser Photon. Rev. 2009; 3(1–2): 180–202. DOI:10.1002/lpor.200810043.
27. Klar T.A., Jakobs S., Dyba M. et al. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA. 2000; 97(15): 8206–8210. DOI:10.1073/pnas.97.15.8206.
28. Gustafsson M.G.L. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA. 2005; 102(37): 13081–13086. DOI:10.1073/pnas.0406877102.
29. Пучков Е.О. Внутриклеточная вязкость: методы измерения и роль в метаболизме. Биологические мембраны. 2014; 31(1): 3–13. [Puchkov E.O. Intracellular viscosity: methods of measurements and role in metabolism. Biological Membranes. 2014; 31(1): 3–13. (In Russ.).] DOI:10.7868/80233475513050149.
30. Puchkov E. Microfluorimetry of Single Yeast Cells by Fluorescence Microscopy Combined with Digital Photography and Computer Image Analysis. Advances in Medicine and Biology. L. V. Berhardt (ed.). N.Y., 2016; 98: 69–90. DOI:10.1134/S0026365619010130.
31. Ohtani M., Saka A., Sano F. et al. Development of image processing program for yeast cell morphology. J. Bioinform. Computat. Biol. 2004; 1(4): 695–709. DOI:10.1142/s0219720004000363.
32. Negishi T., Nogami S., Ohya Y. Multidimensional quantification of subcellular morphology of Saccharomyces cerevisiae using CalMorph, the high-throughput image-processing program. J. Biotechnol. 2009; 141(3–4): 109–117. DOI:10.1016/j.jbiotec.2009.03.014.
33. Nogami S., Ohya Y., Yvert G. Genetic complexity and quantitative trait loci mapping of yeast morphological traits. PLoS Genet. 2007; 3(2): e31. DOI:10.1371/journal.pgen.0030031.
34. Gebre A.A., Okada H., Kim C. et al. Profiling of the effects of antifungal agents on yeast cells based on morphometric analysis. FEMS Yeast Res. 2015; 15(5): fov040. DOI:10.1093/femsyr/fov040.
35. Bjerling P., Olsson I., Meng X. Quantitative live cell fluorescence-microscopy analysis of fission yeast. J. Vi. Exp. 2012; 59: e3454. DOI:10.3791/3454.
36. Akamatsu M., Lin Y., Bewersdorf J. et al. Analysis of interphase node proteins in fission yeast by quantitative and superresolution fluorescence microscopy. Mol. Biol. Cell. 2017; 28(23): 3203–3214. DOI:10.1091/mbc.E16-07-0522.
37. Arasada R., Sayyad W.A., Berro J. et al. High-speed superresolution imaging of the proteins in fission yeast clathrin-mediated endocytic actin patches. Mol. Biol. Cell. 2018; 29(3): 295–303. DOI:10.1091/mbc.E17-06-0415.
38. Bestul A.J., Yu Z., Unruh J.R., Jaspersen S.L. Molecular model of fission yeast centrosome assembly determined by superresolution imaging. J. Cell Biol. 2017; 216(8): 2409–2424. DOI:10.1083/jcb.201701041.
39. Wollman A., Hedlund E.G., Shashkova S. et al. Towards mapping the 3D genome through high speed single-molecule tracking of functional transcription factors in single living cells. Methods. 2020; 170: 82–89. DOI:10.1016/j.ymeth.2019.06.021.
40. Elowitz M.B., Surette M.G., Wolf P.E. et al. Photoactivation turns green fluorescent protein red. Curr Biol. 1997; 7(10): 809–812. DOI:10.1016/s0960-9822(06)00342-3.
41. Uphoff S., Reyes-Lamothe R., de Leon F.G. et al. Single-molecule DNA repair in live bacteria. PNAS. 2013; 110(20): 8063–8068. DOI:10.1073/pnas.1301804110.
42. Virant D., Turkowyd B., Balinovic A. et al. Combining primed photoconversion and UV-photoactivation for aberration-free, live-cell compliant multi-color single-molecule localization microscopy imaging. Int. J. Mol. Sci. 2017; 18(7): 1524. DOI:10.3390/ijms18071524.
43. Yao Z., Carballido-Lуpez R. Fluorescence imaging for bacterial cell biology, from localization to dynamics, from ensembles to single molecules. Annu. Rev. Microbiol. 2014; 68: 459–476. DOI:10.1146/annurev-micro-091213-113034.
44. Stracy M., Kapanidis A.N. Single-molecule and super-resolution imaging of transcription in living bacteria. Methods. 2017; 120: 103–114. DOI:10.1016/j.ymeth.2017.04.001.
45. Gahlmann A., Moerner W.E. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat. Rev. Microbiol. 2014; 12(1): 9–22. DOI:10.1038/nrmicro3154.
46. Uphoff S. Super-resolution microscopy and tracking of DNA-binding proteins in bacterial cells. Methods Mol. Biol. 2016; 1431: 221–234. DOI:10.1007/978-1-4939-3631-1_16.
47. Li Y., Schroeder J.W., Simmons L.A. et al. Visualizing bacterial DNA replication and repair with molecular resolution. Curr Opin Microbiol. 2018; 43: 38–45. DOI:10.1016/j.mib.2017.11.009.
48. Schneider J.P., Basler M. Shedding light on biology of bacterial cells. Phil. Trans. R. Soc B. 2016; 371(1707): 20150499. DOI:10.1098/rstb.2015.0499.
49. Kentner D., Sourjik V. Use of fluorescence microscopy to study intracellular signaling in bacteria. Annu. Rev. Microbiol. 2010; 64: 373–390. DOI:10.1146/annurev.micro.112408.134205.
50. Choi H., Rangarajan N., Weisshaar J.C. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microbiol. 2016; 24(2): 111–122. DOI:10.1016/j.tim.2015.11.004.
51. Haas B.L., Matson J.S., DiRita V.J. et al. Imaging live cells at the nanometer-scale with single-molecule microscopy, obstacles and achievements in experiment optimization for microbiology. Molecules. 2014; 19(8): 12116–12149. DOI:10.3390/molecules190812116.
52. Endesfelder U. From single bacterial cell imaging towards in vivo single-molecule biochemistry studies. Essays Biochem. 2019; 63(2): 187–196. DOI:10.1042/EBC20190002.
1 Другие статьи в «Природе» о люминесценции см.: Лабас Ю.А., Гордеева А.В. Свет и цвет живых организмов (2003. №2. C. 25–31; №3. C. 33–43); Ямпольский И.В., Царькова А.С., Дубинный М.А. и др. Биолюминесценция: возрождение (2014. №7. С. 10–16); Болотин Б.М. Перья жар-птицы (2017. №3. С. 3–12).
2 Линия эпителиальных клеток, полученная из яичника китайского хомяка (англ. Chinese Hamster Ovary, CHO), используется в биологических и медицинских исследованиях, а также для промышленного производства рекомбинантных терапевтических белков. — Примеч. ред.
3 Интерфазные узелки — мультибелковые комплексы в плазматической мембране, которые сливаются вместе и созревают в предшественники сократительного кольца, разделяющего клетку на две в конце митоза (на стадии цитокинеза). — Примеч. ред.
































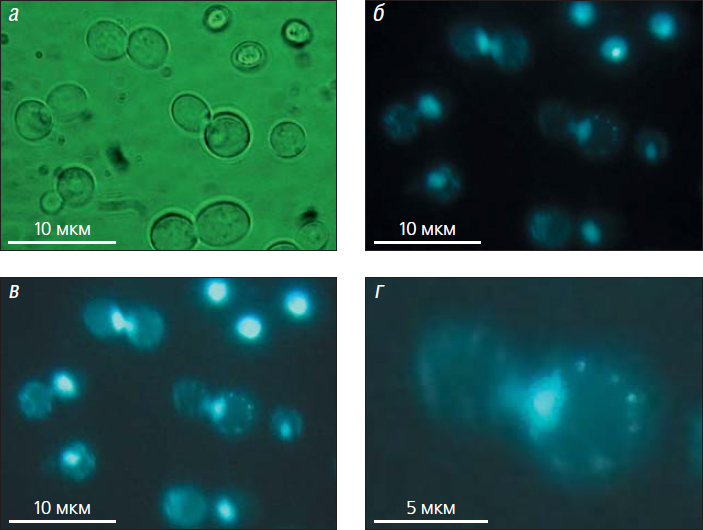
Рис. 1. Компьютерная обработка флуоресцентного изображения для улучшения визуального восприятия: а — режим светопропускания; б–г — режим флуоресценции; в и г — компьютерное улучшение контрастности и увеличение, соответственно. Микроскопия клеток дрожжей Saccharomyces cerevisiae после обработки флуоресцентным красителем нуклеиновых кислот ДАФИ (4’,6-диамидино-2-фенилиндол). После компьютерной обработки стали хорошо видны митохондрии (мелкие точки) и момент деления ядра при образовании дочерней клетки