Ренессанс p-элементов
Аркадий Курамшин,
кандидат химических наук
«Химия и жизнь» №3, 2017

В ХХ веке элементы главных подгрупп Периодической системы были менее популярны, чем те, что расположены в побочных подгруппах. Литий, бор и германий оказались в тени своих дорогих соседей — золота, палладия, родия и платины. Конечно, нельзя не признать, что классические химические свойства элементов главных подгрупп не могут сравниться с быстрыми и элегантными процессами, в которых участвуют комплексы переходных металлов (за открытие этих реакций присуждена не одна Нобелевская премия). В начале 1970-х годов среди химиков вообще бытовало мнение, что элементы главных подгрупп уже раскрыли все свои секреты, а их изучение — фактически пустая трата времени.
Скрытый химический переворот
Когда автор этой статьи был студентом (а он получил диплом Казанского университета в 1992 году), ему и многим его однокурсникам химия p-элементов казалась самым скучным разделом. (Напомним, что s-, p- и d-элементы — это те, валентные электроны которых занимают соответственно s-, p- и d-орбитали.) Нам рассказывали, в какой форме эти элементы существуют в земной коре, преподавали методы их выделения, физические свойства, типичные степени окисления, химические свойства и практическое применение. Вдвойне скучно было тем, кто прошел через химические олимпиады и узнал всю эту полезную информацию еще школьником. Может быть, поэтому в наше время кафедра неорганической химии была не очень популярна при выборе специализации — все мы старались попасть к органикам или элементоорганикам, где рассказывали про наступившую в химии эру переходных металлов, катализирующих все мыслимые и немыслимые превращения веществ.
Тогда еще не было компьютеров и Интернета, всю информацию мы получали только из реферативных журналов по химии и некоторых иностранных журналов, которые выписывала наша библиотека. Ни мы, ни наши преподаватели не знали, что в конце 1980-х годов уже стали заметными первые признаки ренессанса химии элементов главных подгрупп. Именно тогда обнаружили, что возможно получить экзотические формы p-элементов — кремний и фосфор в низкокоординированном и низкоокисленном состояниях, но при этом способные образовывать вполне устойчивые при комнатной температуре соединения. Хотя об их практическом применении в тот момент речь не шла, первые успешные примеры синтеза этих веществ показывали, что химию элементов главных подгрупп немного недооценили и, возможно, придет время, когда p-элементы смогут выйти из тени d- и даже f-элементов. В итоге так и получилось.
Точкой начала разворота к элементам главных подгрупп можно считать 1981 год. Тогда было опубликовано целых три работы, опровергающих представление о том, что устойчивая двойная или тройная связь может образовываться только в том случае, если один из партнеров этой химической связи (а лучше оба) — элемент второго периода. Первым это «правило двойных связей» опроверг Роберт Уэст из университета Висконсина, в группе которого впервые синтезировали устойчивый силен — соединение с двойной связью кремний-кремний, более тяжелый аналог алкенов, знакомых каждому по органической химии (Science, 1981, 214, 4527, 1343–1344, doi: 10.1126/science.214.4527.1343). Вскоре после этого исследователи из Токийского университета, работавшие под руководством Масааки Ёсифудзи, сообщили о синтезе соединения с двойной связью фосфор-фосфор (Journal of the American Chemical Society, 1981, 103, 15, 4587–4589; doi: 10.1021/ja00405a054). В том же году Герд Беккер из университета Штутгарта смог получить устойчивый фосфаалкин — соединение с тройной связью фосфор-углерод, который можно рассматривать как фосфорсодержащий аналог нитрилов карбоновых кислот (Zeitschrift für Naturforschung B, 1981, 36, 16).
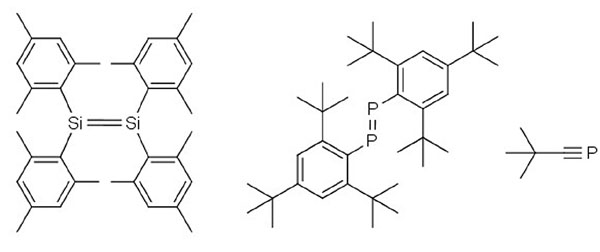
Синтез этих трех соединений вновь подогрел интерес к химии элементов главных подгрупп
Фосфор и кремний — элементы третьего периода, поэтому никто не ожидал от них таких возможностей. В последнем соединении атом фосфора координационно ненасыщен, а это позволяло надеяться, что оно или его аналоги найдут применение в качестве катализаторов. Повод для надежды давало то, что основная задача катализатора — связаться с молекулой-субстратом, которую нужно активировать, на это способны только те молекулы, к которым реагент может легко приблизиться, а в привычных большинству химиков фосфатах атом фосфора, окруженный четырьмя группировками, никак нельзя назвать доступным центром.
Главное — объемное окружение
Все три синтеза, опубликованные в 1981 году, удались потому, что были правильно подобраны заместители, окружающие элементы главных подгрупп в их новых, экзотических соединениях (в химии переходных металлов заместители назвали лигандами). Новые производные, полученные Уэстом, Ёсифудзи и Беккером, объединяло одно — объемные лиганды, связанные с элементами главных подгрупп, стабилизировали кремний или фосфор в низкокоординированном состоянии, которое не было бы устойчивым в других обстоятельствах. Объемные заместители защищают кремний и фосфор от кислорода и воды воздуха, а также не дают вступить в реакцию диспропорционирования и принимать типичные для них степени окисления (+4 и +5 для кремния и фосфора соответственно) и координационные числа (четыре для обоих элементов). Так, силен был стабилизирован четырьмя объемными мезитильными группами (мезитил — это 1,3,5-триметилбензол), а фосфаалкин — объемным трет-бутильным заместителем.
Как только стало ясно, что объемные лиганды делают устойчивыми соединения, в которых p-элементы находятся в невысокой степени окисления и/или с низким координационным числом, к получению новых, необычных производных элементов главных подгрупп стали подключаться и другие ученые. Начиная с 2000-х годов практически в каждом номере Science (а с появления в 2009 году журнала Nature Chemistry — в почти каждом его номере) сообщается о каком-то экзотическом соединении с элементом главных подгрупп.

Этот силилен устойчив, хотя кремний в нем двухвалентный и двухкоординированный Dipp — 2,6-диизопропилфенил
Так, до недавнего времени никто не мог подумать, что удастся получить и охарактеризовать стабильные силилены — кремнийсодержащие эквиваленты карбенов.
Карбены — это частицы с высокой реакционной способностью, в которых на двухвалентном и двухкоординированном атоме углерода либо есть пара электронов (более устойчивый синглетный карбен), либо два отдельных неспаренных электрона (более активный триплетный карбен). В 2012 году Камерон Джонс из австралийского Университета Монаша и его коллеги из Оксфорда и Университетского колледжа Лондона описали первый синглетный силилен — двухвалентный кремний в нем стабилизирован объемным борсодержащим лигандом (Journal of the American Chemical Society, 2012, 134, 15, 6500–6503, doi: 10.1021/ja301042u). Силилен можно выделить в кристаллическом состоянии, и он, заметим, сохраняет устойчивость при температуре до 130°C. А вот в растворе кремниевый аналог карбена димеризуется с образованием силена либо внедряется в C—H связи алканов, воспроизводя химические свойства своих аналогов карбенов.
Химики продолжают получать все новые органические соединения с элементами главных подгрупп. В частности, пытаются заменить в хорошо известной структуре элемент второго периода на аналогичный ему элемент более старшего периода (в этом номере «Хемоскопа» рассказывается о получении фосфорсодержащего аналога одного из первых синтезированных органических веществ). Другое направление немного похоже на коллекционирование редких марок, только вместо марок — химические структуры. Например, в 2016 году Александр Хинц из Оксфорда пытался получить цикл, содержащий атомы четырех различных пниктогенов (элементов 5-й группы главной подгруппы от азота до висмута). Полностью решить задачу ему не удалось — молекула линейного строения не замкнулась в цикл. Тем не менее впечатляет и молекула с уникальной цепью Sb—N—As = P, включающей четыре из пяти p-элементов подгруппы азота (Chemisrty. A European Journal, 2016, 22, 35, 12266–12269, doi: 10.1002/chem.201601916).
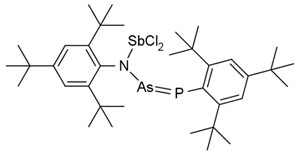
Жемчужина в коллекции химических редкостей — молекула с цепью элементов Sb—N—As = P
Конечно, говорить о синтезе экзотических производных элементов главных подгрупп только как о «химическом коллекционировании» нельзя, поскольку получение аналогов хорошо известных органических соединений, содержащих элементы старших периодов, безусловно важно для уточнения теорий строения химической связи. Разумеется, это не единственная причина интереса химиков. Стремление найти области, в которых эти вещества можно использовать на практике, — именно в нем причина ренессанса химии элементов главных подгрупп.
Еще в 1980-е годы после синтеза первых веществ, в которых наблюдалась низкая координация p-элементов, химики надеялись, что такие координационно ненасыщенные соединения смогут катализировать многие реакции так же, как комплексы переходных металлов. Уж больно заманчиво было бы поменять дорогие соединения платины и палладия на молекулы, содержащие только элементы главных подгрупп. Появившаяся уже в этом тысячелетии информация о свойствах необычных соединений p-элементов подтвердила теоретические прогнозы. Оказалось, что многие из них активируют углеводороды, молекулярный водород и углекислый газ.
Чем плохи переходные металлы?
Казалось бы, зачем разрабатывать новые катализаторы для процессов, которые уже давно отлично ускоряются производными переходных металлов? К тому же и металлоорганическая химия переходных элементов не стоит на месте — все время открываются новые грани реакционной способности d-элементов. Но у благородных переходных металлов есть свои недостатки. В первую очередь цена: самые эффективные катализаторы превращений органических и элементоорганических соединений — это комплексы родия, платины и палладия. Вторая сложность — истощение природных запасов платины и палладия. Наконец еще одна проблема платиновых или палладиевых катализаторов — высокая токсичность. Особенно это актуально при получении лекарств, поскольку их цену существенно повышает стоимость очистки вещества даже от следовых количеств переходных металлов. Переход на новые катализаторы как минимум значительно удешевит лекарственную субстанцию, а возможно, и упростит очистку целевого продукта реакции.
Существуют и дополнительные преимущества, которые может дать применение катализаторов на основе элементов главных подгрупп. Так, не исключено, что некоторые известные реакции пойдут в более мягких условиях, а значит, удастся сэкономить на энергии. Например, еще в 1981 году в своей работе про синтез и свойства первого силена Джонс продемонстрировал, что соединение с двойной связью кремний-кремний может активировать водород при температуре даже меньшей, чем комнатная, тогда как существующие в промышленности процессы гидрирования требуют применения высоких температур.
Один из важных химических процессов, обнаруженных в уже новом тысячелетии, — активация молекулярного водорода с помощью дигермина, германийсодержащего аналога алкинов (Journal of the American Chemical Society, 2005, 127, 12232–12233, doi: 10.1021/ja053247a). Этот процесс, который может показаться обычным, интересен благодаря двум обстоятельствам. Во-первых, несмотря на аналогию строения алкинов и герминов, водород реагирует с последними не по сценарию, характерному для углеводородов с тройной связью углерод-углерод (водород присоединяется к каждому из атомов тройной связи, и гермин превращается в гермен), а по механизму, типичному для атомов переходных металлов. Этот механизм, в результате которого молекула водорода присоединяется к элементу и образуются две новые связи Э—Н (в описанном случае — Ge—H), называется окислительным присоединением и является ключевой стадией многих каталитических процессов с участием переходных металлов. Во-вторых, хотя Н2 и может показаться самой простой и незамысловатой молекулой, химическая связь в ней — самая прочная из всех, какие могут возникать между двумя одинаковыми элементами, поэтому разрыв этой связи и, соответственно, активация водорода в процессах каталитического гидрирования — далеко не простая задача с точки зрения химической технологии.
Можно ли сделать акцептор донором?
Чтобы элемент смог вступить в реакцию окислительного присоединения водорода (независимо от того, где он расположен в Периодической таблице), он должен обладать некоторыми особенностями электронного строения. Процесс Э + H2 = Н—Э—Н пойдет только в том случае, если элемент координационно ненасыщен и его свободная орбиталь может принять электроны молекулярного водорода. Более того, энергия этой свободной орбитали должна быть близка энергии молекулярной орбитали водорода, на которой находятся электроны. Прогресс в области гомогенного металлокомплексного катализа главным образом объясняется тем, что химики, изменяя строение лигандов, связанных с металлом, могут варьировать энергию его орбиталей и таким образом «подстраивать» их под строго определенные вещества, участвующие в реакции. Долгое время считалось, что такая мягкая настройка энергии орбиталей возможна только для d-элементов, однако в последнее десятилетие оказалось, что и для p-элементов тоже. Больше всего надежд исследователи связывают с азотсодержащими комплексами, в которых лиганды, как клешнями, охватывают координационный центр (они и называются хелатирующими лигандами, от лат. chela, клешня), а также со сравнительно новым классом лигандов — N-гетероциклическими карбенами.
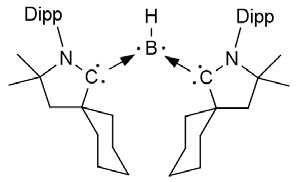
Гетероциклический карбен, полученный Бертраном Dipp — 2,6-диизопропилфенил
Успешный пример применения последних — работа Гая Бертрана из университета Калифорнии в Сан-Диего, в которой эти лиганды стабилизируют атом бора (Science, 2011, 33, 6042, 610–613, doi: 10.1126/science.1207573). Обычно производные бора, содержащего всего три электрона на своем внешнем слое, работают как классический акцептор электронов (кислота Льюиса). Дело в том, что до устойчивой восьмиэлектронной оболочки бору необходимо еще пять электронов, поэтому три ковалентные связи он может образовать из трех своих и трех сторонних электронов, а вот еще два электрона приходится заполучать, принимая в свои пустые электронные ячейки чужую электронную пару. Однако N-гетероциклические карбены — такие сильные доноры электронов, что связанный с ними бор перестает быть акцептором — он становится настолько «электроноизбыточным», что превращается из кислоты Льюиса в основание Льюиса. Еще недавно химики даже не могли спрогнозировать такое значительное изменение свойств хорошо знакомого p-элемента. И хотя работа Бертрана пока интересна только с теоретической точки зрения, переход от теории к практике в наше время происходит довольно быстро.
Далеко ли до катализа
Итак, синтезированные в последнее время производные элементов главных подгрупп могут вступать в ключевые реакции, которые катализируют комплексы переходных металлов. К сожалению, даже упомянутое окислительное присоединение молекулярного водорода к атому кремния или бора — всего лишь первый шаг в последовательности реакций, которые нужно разработать для полного каталитического цикла. Например, если речь идет о гидрировании в присутствии соединений главных подгрупп, механизм которого воспроизводит механизм присоединения водорода в присутствии катализатора Уилкинсона, то после взаимодействия с водородом p-элемент должен образовать комплекс с алкеном, затем должны произойти гидридный перенос и образование комплекса... и все остальные стадии, которые в конечном счете приведут к образованию конечного продукта и регенерации каталитически активной частицы. Только тогда одна частица катализатора даст десятки, сотни или даже тысячи молекул целевого продукта. Но для того чтобы такой каталитический цикл заработал, нужно решить еще много задач — связь элемент-водород, образующаяся в результате окислительного присоединения, не должна быть слишком прочной (а то не произойдет гидридный перенос), элемент, присоединивший водород, должен сохранять низкокоординированное состояние для взаимодействия с алкеном и так далее. Стоит упустить какой-то момент, и катализатора из p-элемента не получится, несмотря на сходство его поведения с d-элементами в некоторых процессах.
Может показаться, что переход от металлокомплексного катализа к катализу соединениями элементов главных подгрупп — слишком сложная задача, и она очень далека от выполнения. Тем не менее интерес к химии p-элементов и желание химиков-синтетиков заменить платиновые или палладиевые катализаторы на что-то другое наверняка обеспечат прорыв в этом направлении. Есть шанс, что мы услышим о катализаторах на основе координационно ненасыщенных элементов главных подгрупп уже в течение ближайшего десятилетия.












Художник Е. Станикова