Методы машинного обучения в биологии
Алёна Таскина, Анна Муравьёва, Алина Ельсукова, Вениамин Фишман
«Природа» №9, 2020
 Алёна Константиновна Таскина — студентка факультета естественных наук Новосибирского государственного университета (НГУ). Область научных интересов — биоинформатика, геномика, эпигенетика; в настоящий момент занимается омиксными исследованиями единичных клеток. |
 Анна Александровна Муравьёва — студентка того же факультета. Область научных интересов — молекулярная и клеточная биология, биоорганическая химия; изучает взаимодействия путей репарации двуцепочечных разрывов ДНК в ранних эмбрионах мыши. |
 Алина Сергеевна Ельсукова — студентка того же факультета, сотрудник Специализированного научно-учебного центра НГУ. Область научных интересов: биоинформатика, геномика и эпигенетика, в частности анализирует последствия хромосомных перестроек. |
 Вениамин Семенович Фишман — кандидат биологических наук, заведующий сектором геномных механизмов онтогенеза Института цитологии и генетики СО РАН, старший преподаватель факультета естественных наук НГУ. Область научных интересов — генетика развития, эпигенетика, геномика и биоинформатика; в последние годы разрабатывает новые математические методы анализа и предсказания укладки хроматина в ядре клетки. |
Современная биология становится наукой «больших данных». Чуть больше десяти лет назад наша лаборатория могла потратить несколько месяцев на определение нуклеотидной последовательности (секвенирование) сотни молекул ДНК. Уже тогда в ходу были первые установки, которые позволяли автоматизировать этот процесс. А сейчас, когда массовое параллельное секвенирование стало рутиной, наша небольшая группа определяет больше миллиарда последовательностей ДНК в год. Раньше львиная доля времени в исследовании уходила на то, чтобы сгенерировать данные, теперь же не меньшую часть времени занимает анализ этих данных.
Количество генерируемых данных в биологии настолько выросло, что для их интерпретации потребовались качественно новые подходы. Раньше типичная постановка эксперимента предполагала, что у исследователя есть заранее сформулированная гипотеза, которую он намерен проверить, и в большинстве случаев проверка гипотезы заключалась в статистическом анализе результатов измерений. Собственно, сама логика статистического анализа предполагает проверку сформулированных a priori гипотез, которые могут быть либо приняты, либо отвергнуты. В случаях, когда получение экспериментальных данных — это долгий и трудоемкий процесс, сформулировать хорошую гипотезу заранее крайне важно, потому что сбор новых данных, необходимых для проверки ортогональной гипотезы, потребует большое количество времени.
Методы, позволяющие в одном эксперименте генерировать гигантские массивы данных, существенно изменили дизайн исследований. Появилась возможность тестировать сразу множество гипотез, а иногда даже формулировать их уже в процессе анализа результатов. Подобная логика эксперимента характерна для современных направлений экспериментальной биологии, называемых омиками (геномики, транскриптомики, протеомики и др.). Омиксные (от англ. суффикса -omics в словах genomics, transcriptomics, proteomics и др.) технологии позволяют проводить количественное исследование большого пула биологических молекул в одном эксперименте [1, 2].
Наша группа исследует, как уложена ДНК в ядре клетки. Раньше для этих целей использовалась цитогенетическая технология 3D-FISH (от англ. fluorescence in situ hybridization — ‘флуоресцентная гибридизация in situ’), основанная на методах микроскопии [3]. Эта технология позволяла в одном эксперименте оценить расстояния только между несколькими заранее выбранными участками ДНК. Поскольку число исследуемых участков сильно ограничено, такой эксперимент требует уже сформулированной гипотезы, которая могла бы объяснить, какие локусы и почему находятся на определенном расстоянии друг от друга. Например, на заре исследований трехмерной организации ядра существовала гипотеза о том, что хромосомы в ядре могут «перемешиваться», и в этом случае участки ДНК, расположенные на разных хромосомах, будут часто располагаться близко друг к другу. Альтернативная гипотеза предполагает существование хромосомных территорий, т.е. отдельных областей, занимаемых каждой хромосомой в ядре клетки. Если эта гипотеза верна, участки ДНК, принадлежащие разным хромосомам, в подавляющем большинстве случаев будут находиться дальше друг от друга, чем участки, расположенные на одной хромосоме. Для того чтобы решить, какая из этих гипотез верна, необходимо заранее определить набор исследуемых локусов: одни из них должны лежать на разных хромосомах, другие — на одной из них. Статистический анализ расстояний между локусами одной или разных хромосом показывает, что верна альтернативная гипотеза, т.е. хромосомы в ядре не перемешиваются, поскольку существует значительная разница в расстояниях между локусами одной и разных хромосом [4].
Сегодня с помощью технологии Hi-C (от англ. high conformation capture — ‘конформации хромосом высокого порядка’), основанной на массовом параллельном секвенировании ДНК, можно исследовать взаимное расположение десятков или сотен тысяч геномных участков в одном эксперименте [5, 6]. А благодаря таким крупным геномным и эпигеномным проектам, как «1000 геномов» (1000 Genomes Project), «Энциклопедия элементов ДНК» (Encyclopedia of DNA Elements, ENCODE) и «Международный нуклеом-консорциум» (International Nucleome Consortium), полученные результаты можно интегрировать с полногеномными данными, ранее опубликованными другими исследователям. Такой материал позволяет тестировать огромное количество гипотез о том, что определяет взаимное расположение тех или иных участков ДНК: их принадлежность определенным хромосомам, взаимодействие ДНК с архитектурными белками или факторами транскрипции, ассоциация хроматина с ядерной ламиной и т.д. Мы можем протестировать сотни гипотез, основываясь на данных, полученных в одном эксперименте.
Теперь гипотезы можно формулировать прямо в процессе анализа данных, но это не означает, что сформулировать хорошую гипотезу стало легче, чем раньше. Наоборот, данных в открытом доступе так много, что человеческий мозг просто не может справиться с объемом информации, увидеть картину целиком и сформулировать разумное предположение о механизмах, определяющих поведение биологической системы. Например, для модельных линий клеток человека доступна информация о вероятности связывания более сотни различных белков с каждым из трех миллиардов нуклеотидов ДНК. Связывание каких из этих белков играет роль в расположении участков ДНК в пространстве ядра и какую именно роль? Что, если важен не отдельный белок, а комбинация белков? В тот момент, когда число параметров биологической системы и их комбинаций становится столь большим, что исключает возможность тестирования роли каждой из них простым перебором, мы входим в область применения методов машинного обучения.
Основная идея подходов машинного обучения заключается в том, что исследователь не должен выбирать заранее гипотезу о том, какие характеристики системы (или какая их комбинация) важна для ее описания. Эти характеристики автоматически выявляются на основе анализа данных. Как именно машина будет искать «удачную» комбинацию параметров, которая описывает состояние системы наилучшим образом, зависит от конкретного метода машинного обучения. Но поскольку возможности перебора разных вариантов у машины существенно больше, чем у человека, такой подход применяется именно в том случае, когда для биологической системы характерно множество сложных, нелинейных зависимостей между разными характеристиками.
Как только закономерности в данных найдены, их можно использовать для предсказания, как поведет себя система при изменении тех или иных параметров. Например, в недавнем исследовании мы показали, что можно предсказать, как изменяется укладка ДНК в клетке после хромосомных перестроек [7, 8]. Для решения этой задачи мы использовали машинное обучение, как способ поиска закономерностей между пространственной укладкой ДНК и различными характеристиками генома в нормальных клетках, а затем, на основе найденных закономерностей, прогнозировали, каким образом будет уложена ДНК, если произойдут мутации.
Область применения и ограничения
Методы машинного обучения могут служить мощным инструментом для решения многих задач, связанных с анализом больших данных. Однако следует отметить один важный недостаток этих методов — они нацелены в первую очередь на поиск закономерностей, а не на построение модели биологического процесса. Закономерности, найденные в ходе машинного обучения, можно эффективно использовать для предсказания поведения системы, но это не означает, что они отражают причинно-следственные связи. К примеру, известно, что сезонное увеличение частоты нападений акул у побережья коррелирует с увеличением продаж мороженого, однако это не означает, что между этими величинами существует причинно-следственная связь. Для алгоритмов машинного обучения она не имеет значения, и это может порождать казусы, подобных ассоциации атак акул с продажей мороженого, публикуемые, в том числе, во вполне уважаемых журналах (один из таких примеров мы подробно разобрали в своей недавней статье [8]). Более того, для некоторых методов машинного обучения в целом сложно извлечь найденные закономерности из построенной модели в интерпретируемой форме. Поэтому, несмотря на всю мощь этих методов для предсказания поведения системы, они зачастую представляют собой «черный ящик», алгоритм действия которого никак не отражает реальный механизм протекающих процессов. Как же методы машинного обучения работают и как их применение может помочь в решении актуальных проблем в самых разных областях биологии?
Предсказание последствий мутаций
Одна из областей биологии, в которой востребованы методы машинного обучения, — геномика. Представим себе, например, следующую задачу из области медицинской геномики. В клинику обращается пациент с наследственным заболеванием. Врачу необходимо поставить пациенту диагноз, указав, какие именно генетические нарушения привели к развитию патологии и, в зависимости от генетической причины патологии и молекулярного механизма ее развития, предложить лечение или коррекцию заболевания. Современные технологии позволяют секвенировать геном пациента [9], но какие из сотен тысяч найденных у пациента генетических вариантов стали причиной заболевания, а какие — просто безобидные «пассажиры», не представляющие никакого вреда? И каков молекулярный механизм развития болезни?
Чтобы ответить на эти вопросы, необходимо, в первую очередь, научиться предсказывать, как каждый из найденных вариантов повлияет на работу генов и структуру кодируемых ими продуктов. Эффекты генетических вариантов, изменяющих белок-кодирующие последовательности, относительно просто предсказать, зная принципы организации генетического кода. Однако на долю таких последовательностей приходится лишь ~2% генома человека. Изменения в других частях генома также могут приводить к развитию патологии, но не за счет нарушения структуры белка, а за счет изменения активности генов и, следовательно, количества кодируемых ими продуктов.
Из-за того, что существует огромное множество различных механизмов, контролирующих экспрессию генов, предсказать последствия изменений в регуляторных участках ДНК крайне сложно. На молекулярном уровне изменение генной экспрессии происходит за счет связывания различных белков с ДНК и их взаимодействия друг с другом. ДНК-связывающих белков много, и их концентрация и активность различается между клетками, и описать эту систему на молекулярном уровне крайне сложно. Однако с точки зрения машинного обучения совершенно не важно, каким именно молекулярным механизмом определяется связывание белка с ДНК и сколько промежуточных посредников участвует в связывании. Машина может вычислить, с какими характерными последовательностями нуклеотидов ДНК белок связывается, а с какими — нет, не отвечая на вопрос о том, почему в одном случае связывание происходит, а в другом — нет. Аналогично, можно определить, как различные комбинации белков влияют на транскрипцию. Тогда, изменив последовательность ДНК в соответствии с найденным генетическим вариантом, можно предсказать, как изменится связывание белков и экспрессия генов.
По приведенной выше логике выстроены работы американских ученых из Принстонского и Гарвардского университетов [10, 11]. Предсказания сайтов связывания различных белков основаны на информации о последовательности нуклеотидов ДНК: для этого исследователи под руководством О. Г. Троянской используют одну из самых популярных технологий машинного обучения — глубокие нейронные сети [12]. Они состоят из слоев, каждый из которых представляет собой отдельный уровень анализа данных. Слой нейросети получает определенную информацию от предыдущего слоя, преобразует ее и передает следующему слою. Самый первый слой получает на входе информацию о последовательности ДНК длиной 1000 букв; последний слой должен выдать информацию о вероятности связывания каждого из 1000 нуклеотидов с интересующим исследователя белком.
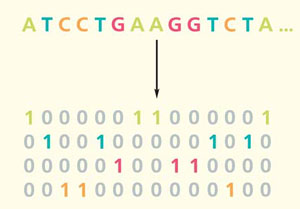
Преобразования, которые проводит каждый из слоев нейросети, сводятся к арифметическим операциям с числами, поэтому последовательность ДНК на входе должна быть представлена в числовом виде. Для этого последовательность ДНК длиной 1000 нуклеотидов представляют в виде четырех массивов из нулей и единиц, каждый из которых содержит 1000 элементов. Каждый из четырех полученных массивов представляет одну букву — А, Т, Г или Ц, и содержит единицы только в тех позициях, в которых соответствующая буква встречается в ДНК (рис. 1). Это одна из наиболее распространенных форм представления последовательности ДНК для машинного обучения, которая называется one-hot-encoding.
В нейросети используются слои трех типов [12]. В слоях первого типа все входные данные разбиваются на перекрывающиеся интервалы длиной восемь элементов. Например, каждый из восьми входных сигналов будет представлять одну букву и кодироваться четырьмя числами по системе one-hot-encoding. Для рассматриваемого интервала все восемь входных сигналов складываются с определенными коэффициентами, в результате получается одно число. Поскольку последовательность из восьми входных сигналов превращается на выходе в одно число, подобная технология называется сверткой (или конволюцией), и построенные по этому принципу нейросети — сверточными, или конволюционными (рис. 2).
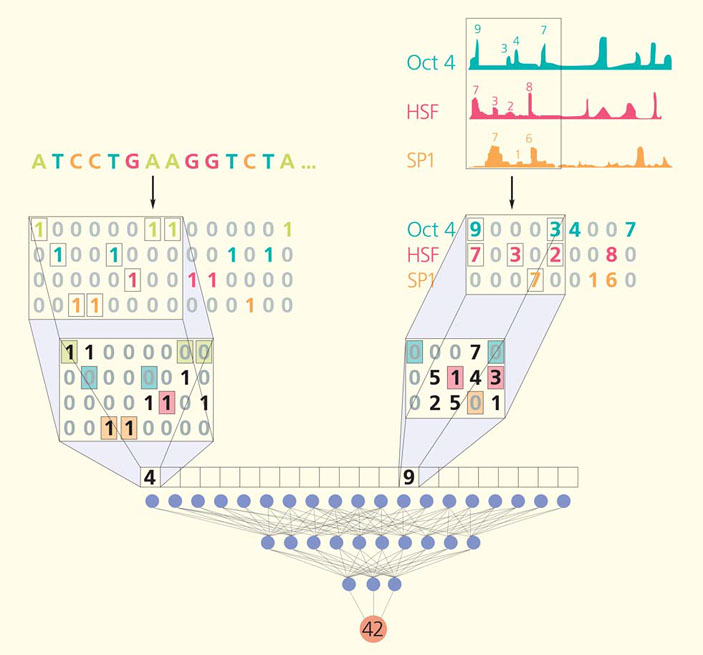
Рис. 2. Сверточная (конволюционная) нейросеть. Входные данные — последовательность букв в определенном участке генома и вероятности связывания белков с ним. На первый слой нейросети данные подаются в числовом виде: последовательность букв представляется в формате one-hot-editing, а информация о связывании отдельного белка в виде строки, в которой каждая цифра отражает высоту пика сигнала Chip-seq в данном месте. Входные данные разбиваются на перекрывающиеся интервалы длиной восемь и пять цифр соответственно. Все входные сигналы из выделенного интервала проходят через фильтр — складываются с определенными коэффициентами; в результате получается одно число. Полученные числа также несколько раз складываются с определенными коэффициентами: отдельные цифры показаны кружочками, процесс сложения с определенными коэффициентами — соединяющими их линиями. Число в розовом круге показывает вероятность связывания белка, для которого проводилось предсказание, с данным участком генома
В зависимости от коэффициентов, одна и та же последовательность сигналов на входе может быть свернута в совершенно разные выходные сигналы (см. рис. 2). Определенный набор коэффициентов, называемый фильтром, позволяет распознать в последовательности один заданный паттерн. В рамках одного слоя сверточной нейросети последовательность анализируют с использованием сотен разных фильтров — с биологической точки зрения это означает, что полученную последовательность проверяют на наличие разных паттернов. Затем полученные паттерны анализируются следующим конволюционным слоем по такому же принципу, но уже с новым набором фильтров.
Суть работы конволюционных слоев удобно представить себе на примере анализа графических изображений — собственно, именно для этой задачи и были первоначально разработаны конволюционные нейросети, и лишь относительно недавно такие модели начали активно применять в биологии. Например, рассмотрим работу нейросети, задача которой — распознать изображение животного на фотографии. Первый слой конволюции ищет на картинке простые объекты: один фильтр выявляет вертикальные линии (такие участки изображения, в которых сумма интенсивностей пикселей, расположенных друг под другом, большая), второй — горизонтальные линии, третий — окружности и т.д. Затем, информация о найденный простых объектах передается в следующей слой, в котором их агрегируют в более сложные паттерны другим набором фильтров, позволяя распознавать, например, отдельные части тела животного: ухо (две диагональные и горизонтальная линия), глаз (две окружности), лапа и т.д.
После нескольких конволюционных слоев, как правило, образуется огромное количество данных. Вернемся к задаче анализа последовательности ДНК. Рассматриваемые 1000 букв можно разделить на 993 частично перекрывающиеся 8-буквенные подпоследовательности, и, если для каждой из них применить 320 фильтров, как сделали авторы статьи [12], получится около 300 тыс. значений. При этом очевидно, что соседние последовательности практически полностью пересекаются, и, вероятнее всего, будут давать похожий результат при анализе одним и тем же фильтром. Поэтому после каждого конволюционного слоя внедряют слой второго типа — субдискретизационный слой (англ. pooling). В этом слое значения, полученные для нескольких соседних участков одним и тем же фильтром, объединяют — либо усредняют их, либо выбирают максимальное из полученных значений.
Конволюционный и субдискретизационный слои позволяют выделить отдельные элементы, присутствующие в небольших участках ДНК (или фрагментах изображения). После этого вся полученная информация анализируется последним слоем нейросети, который называется полносвязным. Этот слой суммирует все полученные на предыдущем слое числа друг с другом с определенными коэффициентами. С биологической точки зрения, конволюционные слои позволяют находить все более и более сложные паттерны в отдельных участках длинной последовательности, а полносвязный слой дает возможность учесть взаимосвязь найденных паттернов между собой. С точки зрения анализа изображений, полносвязный слой рассматривает изображение целиком, а не по отдельным фрагментам, но «видит» его уже не как набор исходных пикселей, а как набор паттернов, выявленных предыдущими слоями. Например, полносвязный слой «видит» на картинке животного: не точки и палочки, а уши, лапы и глаза, и принимает решение, расположены ли эти паттерны на характерных для данного животного расстояниях друг от друга.
Мы привели описание конволюционной нейросети в качестве примера, поскольку подобная архитектура часто используются при анализе геномных данных. Существует множество других архитектур нейросети. Главная задача в обучении любой из них — это выбор коэффициентов, используемых при сложении входных параметров. Для подбора этих коэффициентов необходима обучающая выборка — набор входных данных, для которых известны правильные выходные значения. Коэффициенты подбирают таким образом, чтобы свести к минимуму различия между полученными нейросетью значениями и выходными данными.
Описание нейросетей, приведенное нами, сильно упрощено: например, мы не затрагивали функции активации, применяемые к сигналам при передаче с одного слоя на другой, функции оценки ошибки, методы поиска оптимальных коэффициентов, проблему переобучения нейросетей и многие другие нюансы. Для ознакомления с этими и другими вопросами рекомендуем читателю обратиться к специализированными обзорам.
Натренированная (обученная) нейросеть способна выдавать предсказания даже для тех входных параметров, которые никогда не встречались при обучении. Например, используя конволюционную нейросеть DeepSee, обученную на известных экспериментальных данных о сайтах связывания различных белков, можно предсказывать эффекты мутаций в последовательности ДНК — как ранее описанных, так и никогда не встречавшихся у людей [12].
Используя информацию о связывании белков, полученную при помощи нейросети DeepSee, авторы работы [12] предложили предсказывать экспрессию генов. Для этого также был использован еще один метод машинного обучения, хорошо знакомый биологами — линейная регрессия. Таким образом, на основе двух методов машинного обучения — нейросети, обученной предсказывать связывание белков с ДНК на основе нуклеотидной последовательности, и линейной регрессии, способной предсказывать экспрессию на основе распределения сайтов связывания белков вокруг промотора, можно, зная изменения в последовательности ДНК, предсказать изменения экспрессии генов. Обучив модели на данных нормального генома, авторы предсказали эффекты нескольких тысяч мутаций в различных типах клеток человека, собранных в базе данных GTEx. Для подавляющего большинства из этих мутаций предсказание изменений в экспрессии оказалось точным.
Авторы использовали разработанную модель для анализа эволюции промоторных и регуляторных областей. Они показали, что многие мутации не только уменьшают, но и увеличивают активность промотора, причем наибольший эффект мутаций ожидается в случае эволюционно-консервативных регуляторных элементов. Иначе говоря, высоко консервативные последовательности удерживаются отбором, как правило, в состоянии минимума или максимума активности, и любое движение в сторону от этого состояния вызывает сильное изменение экспрессии. Авторы показали, что мутации в подобных районах часто встречаются у пациентов с различными генетическими заболеваниями и редко — у здоровых людей.
Нейросети, которые предсказывают влияние геномных вариантов на уровень активности генов, могут существенно облегчить процесс диагностики наследственных заболеваний. Однако остается вопрос: как интерпретировать изменения экспрессии? Для некоторых генов экспрессия может варьировать в широких пределах, в то время как для других даже небольшие изменения активности оказываются фатальными. Ответить на этот вопрос тоже помогают методы машинного обучения, которые используют информацию об эволюционной консервативности генома для предсказания патогенности того или иного варианта.
За миллионы лет эволюции геном человека видоизменялся под давлением очищающего отбора, поэтому, как предполагается, варианты, сохранившиеся и распространившиеся в человеческой популяции со времен жизни последнего общего предка с обезьяной, по большей части доброкачественные или нейтральные [13]. Если смоделировать ход эволюции с эквивалентным количеством мутаций, но без влияния отбора, можно получить набор потенциально вредоносных генетических вариантов. Так можно получить две выборки — нейтральных (реально существующих в популяции) и патогенных (не встречающихся у большинства людей) вариантов для тренировки математической модели, позволяющей оценить вероятность генетического варианта быть причиной болезни или же «безобидным пассажиром».
По каким признакам патогенные варианты могут отличаться от непатогенных? Это и признаки, позволяющие предсказать транскрипционный ответ, и эволюционная консервативность последовательности ДНК в районе генетического варианта, отражающая важность изменения транскрипции.
В первую очередь была составлена матрица аннотаций человеческого генома, на основе данных проекта ENCODE, в котором описан большой набор эпигенетических характеристик разных типов клеток человека. Матрицу составили из вариантов генома человека с частотой встречаемости 95–100% по популяции, но при этом отсутствующих у предполагаемого генома общего предка обезьяны и человека по версии Ensembl EPO [14] (рис. 3). Аннотации выбранных вариантов покрывали большое количество типов данных, например, чувствительность к ДНКазе, транскриптомную информацию и меру консервативности.

Рис. 3. Алгоритм обучения логистической модели. Розовым цветом обозначены генетические варианты, которые возникли после расхождения эволюционных путей человека и шимпанзе и остались фиксированными в человеческой популяции. Зеленым цветом обозначены симулированные генетические варианты
Авторы также смоделировали варианты, которые в популяции человека не наблюдали — получилась матрица, содержащая 29,4 млн аннотаций — наполовину наблюдаемых вариантов, наполовину вариантов, построенных на основе моделирования.
Следующий этап обработки данных был основан на применении методов машинного обучения «с учителем» для разделения композиции на вредоносные или потенциально патогенные варианты и безвредные. В последних версиях модели используется метод логистической регрессии.
Построение логистической модели сопряжено с решением задачи классификации объектов. Алгоритм учитывает особенности аннотации генетического варианта и в итоге выдает вероятность принадлежности к вредоносным вариантам или же — простым «пассажирам». Но каким образом набор из множества числовых данных переводится в одно-единственное число между нулем и единицей?
Обучение модели «с учителем» подразумевает, что уже есть композиция из объектов, разделенных на классы [15] — это «правильные» ответы, которые алгоритм должен научиться выдавать. В нашем случае в качестве такой композиции выступает совокупность реальных и модельных вариантов.
Каждому варианту соответствует набор характеристик — меток аннотации: консервативность, чувствительность к ДНКазе и др. Значение каждой характеристики взаимосвязано со степенью вредоносности варианта. Но какие из них имеют решающий голос при классификации, а какие влияют не столь сильно? Именно для ответа на этот вопрос используется вектор весов — набор чисел, по одному для каждой особенности. Чем больше вес, тем более весомый голос приобретает характеристика в глазах алгоритма. В процессе обучения происходит подбор вектора весов таким образом, чтобы разделение вредных и не вредных вариантов было максимально близким к правильному.
С момента своего появления в 2014 г. алгоритм CADD (от англ. Computer Aided Design and Drafting), основанный на уже описанном подходе, стал одним из наиболее широко используемых инструментов для оценки генетической изменчивости человека, а другие инструменты часто используют CADD для оценки их эффективности.
Геномное редактирование можно сделать более безопасным
Итак, с помощью методов машинного обучения можно предсказать влияние тех или иных вариантов на экспрессию генов. Но можно ли исправить вариант, который вызвал заболевание? Современные методы геномного редактирования открывают заманчивую перспективу в этой области. Самый популярный из них — технология CRISPR/Cas9. И в этой области тоже находят применение методы машинного обучения.
В наши дни система CRISPR/Cas9 — революция в мире геномного редактирования. Обычно для изменения последовательностей генов используется комплекс из белка Cas9 и направляющей РНК (guide RNA, или гРНК). Cas9 — это фермент, способный вносить разрыв в ДНК. А гРНК задает специфичность связывания Cas9 с ДНК, «направляя» белок к комплементарной гРНК последовательности. Главное достоинство CRISPR/Cas9 заключается в том, что, используя дизайн гРНК, можно обеспечить специфичность доставки Cas9 почти к любому выбранному району генома. После разрыва обычно происходят модификация и репарация ДНК клеточными системами. Часто для модификации гена используют молекулу-матрицу. Она содержит в себе целевую последовательность, которая в процессе репарации по механизму гомологичной рекомбинации «копируется» в ген в месте разрыва. Это позволяет исправить нужный ген. Именно поэтому большие надежды на CRISPR/Cas9 возлагают в области лечения генетических заболеваний.
Но редактирование генома можно провести и без введения в клетку экзогенной матрицы. Например, концы ДНК после разрыва могут соединиться друг с другом без матрицы при наличии небольшого участка микрогомологии — несколько букв на концах разрыва, комплементарных друг другу. Последовательность ДНК, образовавшаяся в результате микрогомологичного соединения, будет зависеть от длины и расположения участков микрогомологии. Часто репарация происходит без учета микрогомологии — по механизму негомологичного соединения концов — результат репарации по этому пути будет отличаться от микрогомологичного соединения. Существуют и другие альтернативные пути репарации.
Разнообразие путей репарации вносит элемент непредсказуемости в работу системы Cas9 — в зависимости от того, какой из путей репарации реализуется в клетке, образуется один из нескольких возможных генотипов. Наиболее предсказуемым является редактирование с использованием матрицы, однако до недавнего времени использование матрицы было возможно только для делящихся клеток, что существенно ограничивало ее применение [16].
Насколько сложно предсказать результат работы системы Cas9, наглядно демонстрирует работа китайского ученого Хе Цзянкуй, который недавно объявил о рождении генно-модифицированных близняшек. Его целью было приобретение девочками Лулу и Нана устойчивости к ВИЧ на стадии эмбрионов. Но его эксперименты были спланированы и проведены с рядом очень серьезных нарушений. У обеих сестер репарация прошла не так, как планировалось: у одной девочки была обнаружена нецелевая мутация нужного гена, а у другой он и вовсе остался без изменений. Помимо этого, в клетках близняшек обнаружили и мутации вне этого гена, что может сказываться на работе организма. Дальнейшую судьбу девочек сейчас предсказать трудно, а Хе стал одним из первых людей в мире, осужденных за геномное редактирование — китайскими властями он был приговорен к трем годам тюрьмы.
Предсказание генотипов, которые образуются в результате работы системы Cas9 и репарации разрывов, открыло бы абсолютно новые возможности в лечении генетических заболеваний. Недавно такое предсказание было успешно выполнено с использованием методов машинного обучения исследователями Массачусетского технологического института.
Для создания обучающей выборки ученые протестировала работу CRISPR/Cas9 почти на двух тысячах участков человеческого генома. Для каждого из участков были проанализированы мутации, образующиеся при введении системы CRISPR/Cas9 без экзогенной матрицы. Подавляющее большинство мутаций, полученных в эксперименте, представляли собой делеции от 1 до 30 нуклеотидов и однонуклеотидные инсерции. Полученные данные были использованы для обучения inDelphi — алгоритма-предсказателя.
Алгоритм inDelphi состоит из двух модулей. Один из них представляют собой нейросеть, наподобие той, о которой мы уже писали. Задача этой нейросети — предсказать, с какой вероятностью в результате работы системы Cas9 образуется делеция определенной длины. Нейросеть получает на вход три числа: длину и GC-состав микрогомологии (влияют на вероятность микрогомологичного соединения концов), а также длину удаленного участка — этот параметр определяет вероятность того, что делеция могла возникнуть за счет негомологичного соединения концов. Как именно эти параметры связаны с вероятностью возникновения делеции, точно неизвестно. Например, вероятность образования делеции по механизму негомологичного соединения концов зависит от биофизических параметров активности множества белков, участвующих в процессе репарации — экзонуклеаз, лигаз, полимераз. Однако экспериментальные данные, полученные авторами, говорят о том, что частота встречаемости делеций, образующихся по этому механизму, обратно пропорциональна их длине и не зависит от нуклеотидного состава в месте двухцепочечного разрыва. Значит, даже не зная всех деталей молекулярного механизма, можно предсказать вероятность образования делеции заданной длины, просто задав соответствие между длиной и частотой встречаемости на основе экспериментальных данных. Для одного параметра — длины делеции — это относительно простая задача, для решения которой не нужны алгоритмы машинного обучения. Однако наблюдаемые в эксперименте частоты образования делеций представляют собой совокупность действия как минимум двух механизмов — микрогомологичного и негомологичного соединения концов, каждый из которых зависит от своего набора параметров. В этой ситуации найти взаимосвязь между входными параметрами и наблюдаемыми в эксперименте частотами существенно сложней, и для подбора математической функции, описывающей эту зависимость, используется нейросеть.
Второй модуль алгоритма inDelphi анализирует целевую последовательность в определенных нуклеотидных позициях и подсчитывает вероятность вставки нуклеотида в ходе репарации. Экспериментальные данные авторов показали, что для репарации разрывов, вызванных системой CRISPR/Cas9, характерна вставка не более чем одного нуклеотида, причем то, какой нуклеотид будет вставлен, зависит от того, какие два нуклеотида ДНК расположены около сайта разрыва. Авторы предположили, что процессы, ведущие к вставке нуклеотида, конкурируют с делециями, поэтому вероятность появления аллеля с инсерцией зависит от суммарной частоты аллелей с различными делециями.
Для вычисления вероятности вставки каждого из четырех нуклеотидов авторы используют алгоритм k-ближайших соседей. Суть этого алгоритма заключается в том, что из экспериментальных данных выбирают k (k в этом случае было равно пяти) последовательностей, наиболее похожих на анализируемую, и усредненное значение частот инсерции нуклеотида, измеренное для этих пяти локусов, используют в качестве предсказания.
Сходство последовательностей определяют по их нуклеотидному составу вблизи сайтов делеций и вероятности их образования. Поскольку существует всего 16 комбинаций динуклеотидных последовательностей, расположенных около сайтов разрыва, а в эксперименте были протестированы тысячи разных последовательностей, выбор похожих по последовательности участков не составляет труда. Кроме последовательностей динуклеотидов, оценивается суммарная вероятность образования делеций различной длины (предсказанная первым модулем inDelphi) — она должна быть близка к вероятности образования делеций в анализируемом районе генома.
После того, как выбраны пять экспериментальных примеров, наиболее похожих на анализируемый район генома, пять частот инсерций, измеренных экспериментально, усредняют и полученное значение используют в качестве предсказания вероятности инсерции нуклеотида в исследуемом локусе.
На основе работы inDelphi было выяснено, что для 5–11% гРНК можно предсказать генотип с вероятностью более 50%. Также inDelphi выбрал патогенные аллели — «сломанные» варианты генов, которые можно отредактировать CRISPR/Cas9 без молекулы-матрицы. Чаще всего мутации этих аллелей вызывали сдвиг рамки считывания в результате вставки или удаления нескольких нуклеотидов. Это приводило к образованию нефункционального белка и болезни.
И хотя в силу сложной и многогранной механики репарации результат удается предсказать лишь с 50% точностью, авторы продемонстрировали, как можно использовать предложенный подход на практике. Они сделали это на примере коррекции мутантного гена HPS1. Мутация представляет собой удвоение небольшого участка ДНК. Она вызывает синдром Германского — Пудлака — рецессивное заболевание, которое характеризуется нарушением свертывания крови и альбинизмом. Этот синдром распространен в Пуэрто-Рико, где его носителями являются около 5% населения. InDelphi предсказал возможность удаление «лишнего» участка ДНК, на основе чего и был отредактирован ген. Аналогичным образом был восстановлен ген ATP7A; он кодирует белок, который участвует в поглощении меди из пищи и передаче ее ионов от клетки к клетке. Мутантный аллель этого гена связан с болезнью Менкес, при которой наблюдаются серьезные патологии нервной системы. Такие больные редко доживают до трех лет. Таким образом, алгоритм, основанный на машинном обучении, становится следующей ступенью в развитии медицины и открывает новые горизонты лечения множества генетических заболеваний.
Машинное обучение для исследования индивидуальных клеток
Приведенные примеры показывают, что анализ генома человека предоставляет нам большие данные, для анализа которых удобно использовать машинное обучение. Но что, если анализировать не один геном, а десятки, сотни или даже тысячи? Тогда объем данных становится огромным, и можно найти еще более широкое применение для методов машинного обучения.
Одна из областей, в которых рутинно анализируется сразу множество геномов, — single cell технологии (индивидуальный анализ ДНК или РНК в каждой клетке рассматриваемой клеточной популяции). Помимо классических методов секвенирования нуклеиновых кислот, в последние годы появились также возможности исследовать в индивидуальных клетках открытый хроматин, расположение нуклеосом, метилирование ДНК [17, 18]. При объединении этих экспериментов объем данных возрастает еще в несколько раз.
Казалось бы, зачем изучать в отдельности каждую клетку? Гораздо проще секвенировать всю ДНК и/или РНК, выделенную из определенной ткани. В действительности морфологически одинаковые клетки могут состоять из нескольких различных популяций. Такая ситуация наблюдается, например, среди фибробластов мыши. При помощи секвенирования РНК единичных фибробластов выяснилось, что среди них можно выделить две популяции. Они отличаются друг от друга уровнем экспрессии белков-циклинов, которые, в свою очередь, играют важную роль в регуляции жизненного цикла клетки. Различия в уровне экспрессии белка не отражаются на морфологических характеристиках клетки, однако вносят большой вклад в процесс ее жизнедеятельности [19].
Рассмотрим подробнее особенности данных, получаемых в ходе секвенирования отдельных клеток. Статистический анализ оперирует понятиями число наблюдений — n и число признаков — k. Когда n << k, применяют байесовскую статистику, когда n ~ k — частотный подход, когда n >> k — машинное обучение [20]. В области исследования отдельных клеток n >> k, так как число исследованных клеток (n) может быть до нескольких миллионов, а число белок-кодирующих генов (k) примерно 20 тысяч [21].
Основные задачи, которые стоят перед машинным обучением в этой области — это создание алгоритмов для разделения анализируемой популяции клеток на субпопуляции и выяснение и выделение ключевых генов, экспрессия которых характерна для каждой из субпопуляций.
Например, в результате стандартного эксперимента по исследованию транскрипции в индивидуальных клетках можно получить информацию об уровне экспрессии десятков тысяч генов в сотнях тысяч клеток. Какие-то гены показывают существенный уровень различий между клетками, в то время как экспрессия других лишь незначительно колеблется от клетки к клетке. Можно ли на основании этих данных выделить среди исследованных клеток субпопуляции, различающиеся по экспрессии?

Рис. 4. Визуализация данных по экспрессии единичных клеток. Точками отмечены клетки, по осям отложены уровни экспрессии трех генов (А, В, С) в анализируемых клетках. Разными цветами обозначены различные популяции единичных клеток, выделенные на основе отличий в экспрессии генов А, В, С
Если бы генов было немного — два или три, мы могли бы наглядно визуализировать различия между субпопуляциями клеток, отложив на осях в двух- или трехмерном пространстве уровень экспрессии каждого исследованного гена, а каждую клетку изобразив точкой в заданной нами системе координат (рис. 4). Но что делать, если исследованных генов тысячи? Человеческий мозг (как и средства визуализации) не предназначен для анализа тысячемерного пространства!
Для визуализации и анализа таких данных применяется подход уменьшения размерности. Как правило, далеко не все из исследованных генов несут важную информацию о различиях между клетками: экспрессия некоторых генов может быть примерно одинакова для клеток. Кроме того, гены, различающие популяции клеток между собой, часто группируются, формируя паттерны. Приведем такой пример: исследуется образец, который состоит из четырех популяций клеток (рис. 5). В этих клетках существенно различается экспрессия генов А, В и С, но эти различия нельзя считать независимыми: в двух популяция экспрессируются гены А и В (ген С молчит), а в двух других — А и С (ген В молчит). Ни один из генов нельзя выбросить из рассмотрения, иначе мы перестанем различать те или иные популяции. Однако можно просуммировать экспрессию всех трех генов с определенными коэффициентами, задав таким образом экспрессию нового «абстрактного» гена ABC. Складывать экспрессию генов можно с разными коэффициентами, варьируя вклад генов А, В или С в общую сумму. В результате вместо трех настоящих генов А, В и С можно использовать два абстрактных гена, экспрессия которых отражает различия между всеми четырьмя популяциями клеток (см. рис. 5).

Рис. 5. Визуализация данных экспрессии: в популяциях, обозначенных синим и черным цветами экспрессируются гены А и В, ген С не экспрессируется; в популяциях, обозначенных зеленым и красным, не экспрессируются гены А и В, экспрессируется ген С. Показан результат проекции данных на оси главных компонент, а также результаты проекций на плоскости С–В, С–А, А–В
Чем больше различающихся по экспрессии популяций клеток можно выделить в анализируемом образце, и чем меньше колебания в экспрессии генов связаны между собой, тем больше понадобится «абстрактных» генов для описания такой системы. При этом небольшими различиями часто можно пренебречь или сделать их вклад в определение уровня экспрессии «абстрактных» генов малозначительным. Задача алгоритмов машинного обучения — задать «абстрактные» гены таким образом, чтобы, с одной стороны, минимизировать их число и, с другой стороны, чтобы различия в экспрессии «абстрактных» генов наиболее полно отражали различия между экспрессией реальных генов в клетках.
Как и в большинстве случаев в биологии, математически такая задача была сформулирована и решена уже давно: с точки зрения математики мы хотим понизить размерность пространства характеристик (генов) с k-мерного до двух- или трехмерного, максимально сохранив различия между образцами (клетками).
Один из использующихся алгоритмов для решения такой задачи — метод главных компонент. С помощью этого метода можно задать «абстрактные» гены таким образом, чтобы дисперсия (мера разброса) между клетками по экспрессии этих генов была максимальна. Если выбрать два «абстрактных гена» и для каждой клетки по оси Х отложить значение экспрессии первого гена, а по оси Y — значение экспрессии второго, можно визуализировать клетки в виде точек на плоскости. Так как мы выбрали абстрактные гены таким образом, чтобы различия в их экспрессии были максимальны, расстояние между клетками-точками будет наиболее правдоподобно отражать суммарную разницу в их экспрессионных профилях.
Рис. 6. Наиболее распространенные архитектуры нейронных сетей. Кружками обозначены отдельные цифры или строки цифр, линиями обозначены операции сложения цифр или строк с определенными коэффициентами (как на рис. 2)
Малое количество образца, выделяемого из одной клетки, также вносит свои особенности в структуру single-cell данных. Нулевые значения, получаемые в результате таких экспериментов, могут быть как истинными, так и ложными. Истинные нулевые значения отражают тот факт, что данный ген не экспрессируется в этой клетке, а ложные получаются в результате потери части анализируемого образца и представляют собой пропуски в данных. Примечательно, что проблема разреженных данных возникает и при построении рекомендательных систем, так как далеко не все пользователи оставляют свои оценки фильмам. Для восстановления пропусков в разреженных данных в машинном обучении существует отдельный класс алгоритмов [22, 23]. Для решения задачи восстановления пропущенных значений используются нейросети автокодировщики. Для обучения автокодировщика в данные об экспрессии единичных клеток вносят пропуски в случайных местах. Полученные строки чисел представляют собой входные параметры для нейросети. Строки, отражающие экспрессию генов, складываются между собой с определенными коэффициентами, полученные значения передаются последующим слоям нейросети. Особенность автокодировщиков состоит в том, что на выходящем слое строк, представляющих экспрессию генов, столько же, сколько и на входящем, а на внутреннем слое их меньше (рис. 6). Коэффициенты нейросети подбирают таким образом, чтобы данные с пропусками, проходя через автокодировщик, как можно лучше совпадали с начальными данными без пропусков. Вначале, на этапе снижения пространства данных, нейросеть находит паттерны коэкспрессии, т.е. выделяет гены, экспрессия которых зависит друг от друга. Затем, на этапе восстановления пространства данных, нейросеть воссоздает зависимость в экспрессии генов из каждого паттерна, при этом и происходит вставка пропущенных значений (рис. 7).
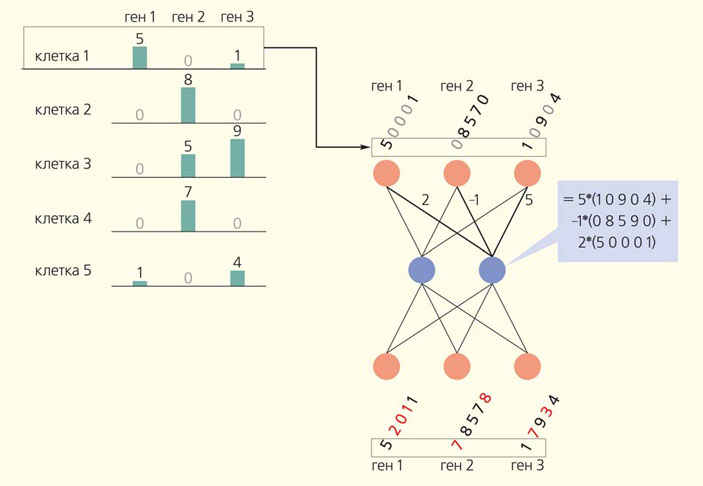
Рис. 7. Восстановление пропущенных значений в данных об экспрессии единичных клеток при помощи нейросети автокодировщик. Входными данными является уровень экспрессии генов в отдельных клетках. Каждый ген представляется в виде отдельной строки, где цифры отражают уровень экспрессии данного гена в определенной клетке. Далее числовые строки складываются с определенными коэффициентами: кружками показаны отдельные строки, а линиями операции сложения. На последнем слое нейросети получаются строки, отражающие экспрессию данных генов в отдельных клетках, при этом на месте пропусков в данных вставлены определенные значения (показаны красным)
Такие нейросети, как автокодировщики, используют и для понижения размерности данных секвенирования единичных клеток [24]. После проведения эксперимента для каждого гена известно, в каких клетках он экспрессируется, а в каких нет. Используя эту информацию, для каждого из генов строится строка, состоящая из единиц и нулей: на местах, соответствующих номерам клеток, в которых присутствует продукт этого гена, ставим 1, а на оставшихся местах — 0. Каждая операция сложения строк друг с другом представляет собой создание «абстрактного гена» — по аналогии с ранее приведенным примером, а коэффициенты задают вклад реально экспрессирующихся генов в «абстрактные». Коэффициенты нейронной сети подбираются таким образом, чтобы значения, выдаваемые выходящим слоем, как можно лучше совпадали со значениями входящего слоя. Таким образом, сначала нейросеть создает из большого числа настоящих генов небольшое число абстрактных, а затем, суммируя абстрактные гены друг с другом, вновь воссоздает из них профиль экспрессии реальных генов. Значения, полученные внутренним слоем автокодировщика, как раз и рассматриваются в качестве пространства сниженной размерности, так как число их меньше, чем число входящих генов. Таким образом, автокодировщики выделяют «абстрактные гены» так, чтобы по ним можно было точно восстановить данные о настоящих генах.
После снижения размерности выполняется задача кластеризации — выделения групп клеток, наиболее похожих друг на друга.
Как и с конволюционными нейросетями, методы снижения размерности были заимствованы биологами из других областей науки и технологий. Например, методы снижения размерности и кластеризации повсеместно используются рекомендательными системами онлайн-кинотеатров. Как каждую клетку можно описать экспрессией генов, так каждого пользователя — оценками просмотренных им фильмов. Затем с помощью упомянутых методов можно выделить группы зрителей, сходных по предпочтениям. Рекомендации строятся на основе того, какие фильмы понравились людям из сходной группы [25].
Благодаря применению методов машинного обучения для анализа единичных клеток стало возможным рисовать атласы органов в одноклеточном разрешении. Создание таких атласов открывает много возможностей в самых разных областях биологии и медицины. Например, в недавнем исследовании нашли на карте клеточных популяций эпителия дыхательных путей отдельный тип клеток — ионоциты, в которых экспрессируется ген CFTR [26]. CFTR кодирует трансмембранный белок, который за счет энергии АТФ транспортирует ионы хлора из клетки наружу. Открытие ионоцитов имеет большое значения для поиска подходов к лечению одного из самых распространенных генетических заболеваний — муковисцидоза, при котором ген CFTR в результате мутации теряет свою функцию транспортировки ионов хлора. Последующее смещение ионного равновесия приводит к нарушению циркуляции жидкости в межклеточном пространстве. При муковисцидозе поражаются многие органы (легкие, кишечник, протоки поджелудочной железы и потовых желез), потому что секрет железистых клеток их эпителия становится слишком густой. Сгущается и легочный сурфактант, который в норме снижает поверхностное натяжение альвеол и защищает их от проникновения инфекций. Густая слизь забивает дыхательные пути, препятствует работе клеток иммунной системы и способствует развитию бактериальных пленок [27]. Для применения подходов генной терапии к лечению муковисцидоза необходима доставка медикамента к тем клеткам, в которых непосредственно экспрессируется ген CFTR. Открытие ионоцитов, сделанное благодаря анализу single-cell данных методами машинного обучения, и их картирование на легких человека открывает новую возможность в лечении муковисцидоза.
***
Основное преимущество машинного обучения заключается в возможности генерировать и проверять множество гипотез на основе больших наборов экспериментальных данных. Алгоритмы, которые применяются для решения биологических задач, как правило, приходят в биологию из других сфер науки о больших данных. Учитывая, насколько интенсивно развивается эта область, можно ожидать, что в ближайшее время использование подходов машинного обучения в биологии будет активно расширяться.
Литература
1. Perez-Riverol Y., Zorin A., Dass G. et al. Quantifying the impact of public omics data // Nat. Commun. 2019; 10(1): 1–10. DOI: 10.1038/s41467-019-11461-w.
2. Karczewski K. J., Snyder M. P. Integrative omics for health and disease // Nat. Rev. Genet. 2018; 19(5): 299–310. DOI: 10.1038/nrg.2018.4.
3. Huber D., Voith von Voithenberg L., Kaigala G. V. Fluorescence in situ hybridization (FISH): History, limitations and what to expect from micro-scale FISH? // Micro Nano Eng. 2018; 1: 15–24. DOI: 10.1016/j.mne.2018.10.006.
4. Cremer T., Cremer M. Chromosome territories // Cold Spring Harb Perspect Biol. 2010; 2(3). DOI: 10.1101/cshperspect.a003889.
5. Wit E. de, Laat W. de A decade of 3C technologies: Insights into nuclear organization // Genes Dev. 2012; 26(1): 11–24. DOI: 10.1101/gad.179804.111.
6. Denker A., Laat W. de The second decade of 3C technologies: Detailed insights into nuclear organization // Genes Dev. 2016; 30(12): 1357–1382. DOI: 10.1101/gad.281964.116.
7. Fishman V. S., Salnikov P. A., Battulin N. R. Interpreting chromosomal rearrangements in the context of 3-dimentional genome organization: a practical guide for medical genetics // Biochemistry (Moscow). 2018; 83(4): 393–401. DOI: 10.1134/S0006297918040107.
8. Belokopytova P. S., Nuriddinov M. A., Mozheiko E. A. et al. Quantitative prediction of enhancer-promoter interactions // Genome Research. 2020; 30(1): 72–84. DOI: 10.1134/S1022795419100089.
9. Mozheiko E. A., Fishman V. S. Detection of point mutations and chromosomal translocations based on massive parallel sequencing of enriched 3C libraries // Russ. J. Genet. 2019; 55(10): 1273–1281. DOI: 10.1134/S1022795419100089.
10. Zhou J., Theesfeld C. L., Yao K. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk // Nat. Genet. 2018; 50(8): 1171–1179. DOI: 10.1038/s41588-018-0160-6.
11. Kelley D. R., Reshef Y. A., Bileschi M. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks // Genome Res. 2018; 28(5): 739–750. DOI: 10.1101/gr.227819.117.
12. Zhou J., Troyanskaya O. G. Predicting effects of noncoding variants with deep learning-based sequence model // Nat. Methods. 2015; 12(10): 931–934. DOI: 10.1038/nmeth.3547.
13. Rentzsch P., Witten D., Cooper G. M. et al. CADD: Predicting the deleteriousness of variants throughout the human genome // Nucleic Acids Res. 2019; 47(D1): D886–D894. DOI: 10.1093/nar/gky1016.
14. Herrero J., Muffato M., Beal K. et al. Ensembl comparative genomics resources // Database. 2016; 2016: baw053. DOI: 10.1093/database/baw053.
15. Tolles J., Meurer W. J. Logistic regression: Relating patient characteristics to outcomes // JAMA. 2016; 316(5): 533–534. DOI: 10.1001/jama.2016.7653.
16. Shen M. W., Arbab M., Hsu J. Y. et al. Predictable and precise template-free CRISPR editing of pathogenic variants // Nature. 2018; 563(7733): 646–651. DOI: 10.1038/s41586-018-0686-x.
17. Butler A., Hoffman P., Smibert P. et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species // Nat. Biotechnol. 2018; 36(5): 411–420. DOI: 10.1038/nbt.4096.
18. Clark S. J., Argelaguet R., Kapourani C. A. et al. ScNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells // Nat. Commun. 2018; 9(1): 781. DOI: 10.1038/s41467-018-03149-4.
19. Mahmoudi S., Mancini E., Xu L. et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing // Nature. 2019; 574(7779): 553–558. DOI: 10.1038/s41586-019-1658-5.
20. Bzdok D., Altman N., Krzywinski M. Points of significance: statistics versus machine learning // Nat. Methods. 2018; 15(4): 233–234. DOI: 10.1038/nmeth.4642.
21. Cao J., Spielmann M., Qiu X. et al. The single-cell transcriptional landscape of mammalian organogenesis // Nature. 2019; 566(7745): 496–502. DOI: 10.1038/s41586-019-0969-x.
22. Badsha M. B., Li R., Liu B. et al. Imputation of single-cell gene expression with an autoencoder neural network // bioRxiv. December 2018: 504977. DOI: 10.1101/504977.
23. Talwar D., Mongia A., Sengupta D., Majumdar A. AutoImpute: Autoencoder based imputation of single-cell RNA-seq data // Sci. Rep. 2018; 8(1): 16329. DOI: 10.1038/s41598-018-34688-x.
24. Wang D., Gu J. VASC: Dimension reduction and visualization of single-cell RNA-seq data by deep variational autoencoder // Genomics, Proteomics Bioinforma. 2018; 16(5): 320–331. DOI: 10.1016/j.gpb.2018.08.003.
25. Symeonidis P. Content-based dimensionality reduction for recommender systems // Studies in Classification, Data Analysis, and Knowledge Organization. 2008; 619–626. DOI: 10.1007/978-3-540-78246-9_73.
26. Plasschaert L. W., Žilionis R., Choo-Wing R. et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte // Nature. 2018; 560(7718): 377–381. DOI: 10.1038/s41586-018-0394-6.
27. Clunes M. T., Boucher R. C. Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder // Drug Discov. Today Dis. Mech. 2007; 4(2): 63–72. DOI: 10.1016/j.ddmec.2007.09.001.














Рис. 1. Числовое представление последовательности ДНК в формате one-hot-editing