От науки к интернет-играм: коллективное решение биоинформатических задач
Михаил Гельфанд
«Природа» №11, 2014
 Об автореМихаил Сергеевич Гельфанд — доктор биологических наук, профессор, член Европейской академии, заместитель директора Института проблем передачи информации им. А. А. Харкевича РАН, профессор факультета биоинженерии и биоинформатики Московского государственного университета им. М. В. Ломоносова. Область научных интересов — биоинформатика, молекулярная эволюция, системная биология, сравнительная и функциональная геномика, метагеномика. |
В биоинформатике есть много задач, которые наиболее эффективно решаются сочетанием компьютерного и экспертного анализа. В методических разделах научных статей этап «сделали вручную» обычно не афишируется, потому что его трудно строго описать, в то же время ручная подгонка, например, выравниваний последовательностей считается совершенно естественной и приемлемой. Однако есть случаи, в которых такая обработка — основной технический прием, причем производится она не группой специалистов, а большим сообществом пользователей Интернета. Это связано с тем, что для многих задач относительно легко построить функционал, оценивающий качество решения, но вычислительно очень сложно его оптимизировать: пространство решений велико и имеет множество локальных оптимумов. Оказалось, что, если оформить задачу поиска оптимального решения как интернет-игру, найдется множество желающих в ней поучаствовать.
Первый проект
Исторически первым проектом такого рода стала игра Foldit (от англ. fold it — ‘сложи это’), направленная на решение традиционной задачи вычислительной биологии и биофизики — предсказание пространственной структуры белка по его аминокислотной последовательности. Экспериментальные методы анализа белковой структуры, кристаллография и ЯМР-спектроскопия, очень трудоемки. Компьютерное же прогнозирование полезно при определении функции белка, поиске взаимодействующих с ним малых молекул (а значит, конструировании лекарств) и анализе белок-белковых взаимодействий. Кроме того, для предсказаний, сделанных с помощью компьютерных программ, уровень соответствия действительности показывает, насколько хорошо мы понимаем механизмы образования пространственной структуры. Все началось с проекта Rosetta@home. Создатели алгоритма Rosetta, который предсказывал пространственную структуру белка ab initio, без учета информации о структурах родственных ему белков, построили такую функцию вычисления свободной энергии, которая, как правило, имела минимум для нативной структуры молекулы [1]. Но, как это часто бывает в биоинформатике, высокое качество функционала привело к сложностям его оптимизации для конкретных белков: вычисления занимали слишком много времени. Тогда авторы обратились к опыту проекта SETI@home, в котором владельцам персональных компьютеров предлагали пожертвовать свободное время своих процессоров для поиска сигналов внеземных цивилизаций. Аналогичная программа распределенных вычислений была разработана и на основе алгоритма Rosetta. Компьютерная заставка показывала текущую структуру белка, и пользователи часто наблюдали, как алгоритм подолгу блуждает в окрестности локального оптимума, не делая очевидных с точки зрения человека шагов (например, таких, как перемещение внутрь молекулы торчащего наружу большого гидрофобного остатка). Дело в том, что разложение этого действия на элементарные составляющие, которыми оперирует алгоритм, требует отклонения в область высоких энергий, так как молекулу надо локально «распутать» (что энергетически невыгодно), переместить гидрофобный остаток, а потом «свернуть» заново. Поэтому, пообщавшись с такими пользователями, авторы проекта решили привлечь их к решению содержательных научных задач.
Для этого был разработан простой веб-интерфейс (рис. 1) с естественным набором элементарных операций. Некоторыми участник игры управлял с помощью компьютерной мыши. Так, можно было перемещать индивидуальный аминокислотный остаток (и связанные с ним остатки — с сохранением стерических ограничений) или целый элемент молекулярной структуры. Другие операции представляли собой упрощенные (для сокращения времени) версии процедур базового алгоритма Rosetta — это глобальная оптимизация свободной энергии либо, наоборот, ее локальная минимизация за счет движений основной цепи в заданном интервале или за счет поворотов боковых остатков. Кроме того, визуальный интерфейс выделял пространственные конфликты между боковыми остатками, которые необходимо разрешить, а также энергетически невыгодные пустоты в структуре и торчащие наружу гидрофобные остатки. Был разработан набор учебных задач, который вводил элементарные операции и давал возможность пользователям понять, как они работают. На веб-сайте проекта регулярно появлялись новые аминокислотные последовательности белков, а за каждую предсказанную структуру в соответствии с абсолютной величиной ее свободной энергии пользователю начислялись игровые баллы.

Видимо, ключевым элементом успеха проекта (рис. 2) стало то, что его авторы не ограничились просто игрой, а создали целое сообщество игроков. Для каждого белка велись записи текущих рекордов, отмечались суммарные достижения конкретного участника за все время игры. Более того, были созданы средства для общения игроков, обмена удачными находками и формирования команд, совместно работающих над новыми задачами. Примечательно, что в числе авторов первой опубликованной работы, посвященной проекту, значится некто под именем Foldit players (игроки Foldit; если точнее — указано, что их более 57 тыс.) [2]. В этой статье сопоставлялись результаты автоматического предсказания пространственной структуры белка с помощью алгоритма Rosetta и прогнозы, сделанные коллективом игроков. Эталоном служили соответствующие структуры, определенные кристаллографическими методами. Во многих случаях результаты, полученные участниками проекта, оказались существенно лучше, чем чисто автоматические (а качество оценивается близостью структуры к нативной). Игроки проявляли себя с наиболее сильной стороны в ситуациях, когда уже есть автоматически предсказанная конформация белка с правильно определенными основными элементами его вторичной структуры и их взаимным расположением и требуется провести доработку, например, убрать торчащие гидрофобные остатки или уточнить ориентацию элементов вторичной структуры. Более тонкая работа по уточнению положений боковых аминокислотных остатков производится компьютером, причем участники неоднократно делают это в процессе игры. Таким образом, успех работы критическим образом зависит от того, удается ли автоматической процедуре угадать общие черты структуры и оказаться в достаточно близкой окрестности оптимальной конформации. К счастью, часто современные алгоритмы способны это сделать. Интересно, что в командах игроков само собой происходит разделение ролей: каждый делает то, что у него лучше получается, — от анализа структуры «с высоты птичьего полета» до уточнения мелких деталей.
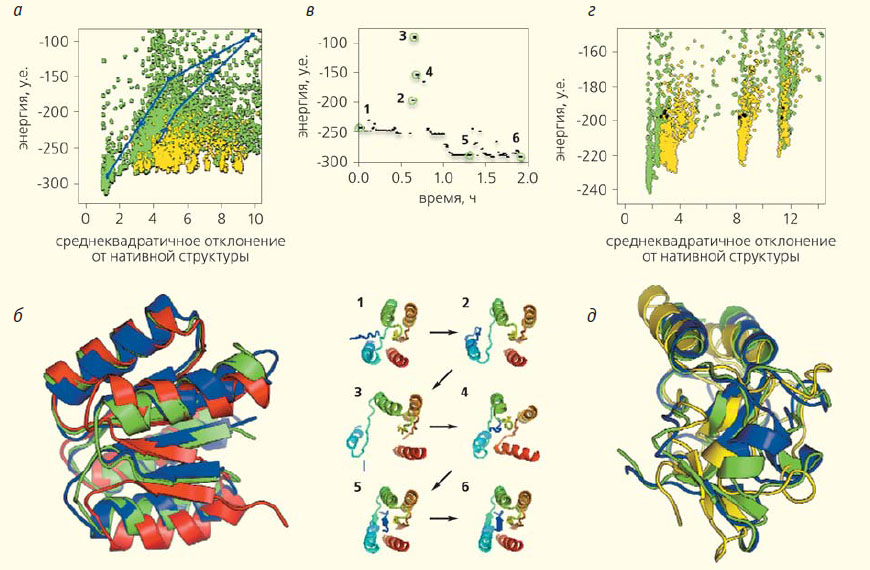
Рис. 2. Примеры задач, в решении которых игроки Foldit превзошли компьютер [2]. Решение задачи № 986875, которая была посвящена предсказанию пространственной структуры белка 2kpo (а—в), и задачи № 986698 о белке 2kky (г, д). Конформации, полученные игроками, показаны зелеными точками, а программой Rosetta — желтыми. По горизонтальной оси отложено полноатомное среднеквадратичное отклонение предсказанной структуры от нативной (опубликованной), по вертикальной — ее энергия (а, г). Черной точкой отмечено начальное приближение (4,28 Å от нативной конформации). Наилучшие результаты Foldit отличаются от нативной структуры на 1,4 Å, а в случае программы Rosetta — на 2 Å. Голубые линии соответствуют траектории одного из игроков (а). Наложение предсказания Foldit (изображено зеленым) на структуру белка 2kpo, экспериментально определенную с помощью ЯМР (синим). Стартовое приближение, в котором концевой тяж переставлен с соседним (и образует неправильную конформацию), показано красным. 8% игроков смогли верно расположить эти тяжи (б). Траектория самого успешного в задаче № 986875 игрока показывает, как он проходил области высоких энергий в поисках оптимальной структуры белка 2kpo (снизу изображены переходы 1–6). По вертикали отложена энергия предсказанной конформации, по горизонтали — истекшее время (в). Сравнение структур, полученных игроками и программой Rosetta для белка 2kky: игрокам удалось выбрать из множества стартовых конформаций (отмечены черным) ближайшую к нативной структуре и улучшить ее (г). Наложение предсказания Foldit (изображено зеленым) на нативную структуру белка 2kky (синим). Стартовое приближение показано желтым (д)
Успехи игроков
Предсказание пространственной структуры белка — область, где очевидны критерии качества (заметим, что не все биоинформатические задачи таковы), а значит, возможно и соревнование. Раз в два года такой конкурс действительно проводится, он называется CASP (Critical Assessment of protein Structure Prediction). Для него выбирают белки, структуры которых уже определены, но еще не опубликованы. Участники делают свои предсказания по аминокислотной последовательности, а потом публикуются и предсказания, и нативные структуры. При этом участники соревнуются в различных категориях, в зависимости от того, какая задача решается (например, можно предсказывать не структуру белка, а то, как он взаимодействует с малыми молекулами) и какие данные доступны. Поскольку для многих белков известны структуры их гомологов, рабочая группа Foldit дополнила набор игровых операций. Теперь участники проекта могли использовать выравнивания аминокислотных последовательностей мишени и белков, конформация которых уже известна [3]. Хотя в категории предсказания белковой структуры по гомологии игроки в целом оказались не очень успешны, одна группа смогла превзойти остальные в анализе одной сложной структуры. Самым же ярким успехом стало предсказание структуры протеазы ретровируса Мейзона-Пфайзера (M-PMV, Mason-Pfizer Monkey Virus) — возбудителя, вызывающего синдром приобретенного иммунодефицита обезьян. Это небольшой белок длиной 114 аминокислот, он уже давно был кристаллизован, но, несмотря на это, определить его структуру не получалось. Игроки нашли конформацию, послужившую хорошим начальным приближением, которое затем удалось улучшить традиционными кристаллографическими методами, и в результате структура белка была определена [4]. При этом в научной публикации виновники торжества решили не указывать свои настоящие имена, а в обсуждении хода работы и конкретных решений, существенно способствовавших достижению общей цели, упоминаются игровые имена игроков [3]. Это необычно для «классического» научного мира, где важно и кто значится в списке авторов, и на каком месте.
Следующий шаг также был сделан в результате наблюдений над совместной работой игроков. Создатели проекта Foldit разработали специальную систему, которая позволяла игрокам делиться «рецептами» — используемыми ими стандартными последовательностями операций [5]. Сначала это делалось с помощью графического интерфейса, а затем был придуман простой язык. Игроки могли модифицировать «рецепты», находящиеся в общем доступе, а разработчики классифицировали их по нескольким категориям (в зависимости от решаемых задач и используемых операций), следя за всем процессом, и собирали статистику. Одновременно с этим шла независимая работа по совершенствованию алгоритма автоматического предсказания для более быстрого, но не менее надежного поиска оптимальной пространственной структуры белка.
Последнее по времени достижение игроков — работа по улучшению фермента, катализирующего реакцию Дильса — Альдера, одну из основных в органической химии. Требовалось не только поменять аминокислотные остатки в функциональном центре фермента, но и структуру самого центра, т.е. ход основной цепи молекулы. Опять пополнился набор инструментов: теперь можно было вставлять аминокислотные остатки и заменять одни на другие. Предсказанные белки химически синтезировали и проверяли их ферментативную активность. Чтобы облегчить взаимодействие между коллективом игроков и экспериментальной лабораторией, один из участников взял на себя роль посредника — формулировал подзадачи, постепенно приближающие всех к желанной цели. Сначала смоделировали изменения в функциональном центре фермента, благодаря которым увеличилось количество его контактов с лигандами, а потом добавили элемент, стабилизирующий пространственную структуру активного центра. В результате был получен фермент, активность которого в 18 раз выше, чем у исходного [6].
Развитие идеи
Естественно, появились и другие проекты, построенные на схожих идеях. Создатели EteRNA описывают свой проект как «большую открытую лабораторию» [7]. Игроки (в феврале 2014 г. их было уже 37 тыс.) предлагают последовательности РНК, которые должны свернуться в заранее указанные организаторами структуры. Каждую неделю голосованием выбирается восемь последовательностей. Их синтезируют в экспериментальной лаборатории, а затем проверяют состояние каждого нуклеотида (спарен он или нет) и тем самым определяют, насколько хорошо подобрана последовательность. Это обратная задача по отношению к более известной — предсказанию вторичной структуры РНК по нуклеотидной последовательности. Участники проекта начинали с того, что пытались использовать компьютерные программы для решения прямой задачи, чтобы подобрать ответ к обратной. Однако синтезированные молекулы сворачивались не так или не только так, как надо было. Постепенно у сообщества накапливался соответствующий опыт, который формализовали в виде все увеличивающегося набора эмпирических правил. Часть из них, связанная с минимизацией симметрии и ролью гуанин-цитозинового состава, уже была известна ученым, изучающим структуры РНК, однако некоторые правила были совсем новыми, например, накладывающими ограничения на использование конкретных нуклеотидов рядом с основаниями шпилек или положение гуанинов в петлях. Структурно-молекулярная основа этих новых принципов до сих пор неясна. Оказалось, что игроки могут не просто подбирать последовательности, принимающие заданную конформацию, но и, как в случае с белками, находить структуру, связывающую заданную молекулу (лиганд).
В задаче построения множественного выравнивания нуклеотидных последовательностей экспериментальная проверка невозможна в принципе, поэтому в проекте Phylo обратные связи между его создателями и игроками практически отсутствуют [8]. Организаторы сделали игру максимально абстрактной: участники выравнивают не нуклеотиды, а разноцветные геометрические фигурки, используя даже сотовые телефоны и планшеты. Задачи при этом решаются настоящие, научные — улучшить 739 проблемных участков во множественном выравнивании регуляторных областей генов позвоночных. Единственная уступка, которую авторы делают биологии, — они позволяют игроку самому выбрать ген, исходя из его медицинской важности. Итак, на наших глазах развертывается замечательный социальный эксперимент, существенно меняющий представления о том, кто и как должен и может заниматься наукой. Участники проектов контактируют друг с другом и с разработчиками, которые внимательно следят за происходящим в игре и постоянно улучшают возможности взаимодействия. За короткое время образовалось три сообщества, насчитывающих десятки тысяч участников, решающих сложные научные задачи и активно обменивающихся идеями, использование которых позволяет совершенствовать существующие алгоритмы. Принципы построения подобных сообществ различны, но общее для них — игровая составляющая. Организаторы формулируют биологические задачи доступно и увлекательно для людей, не специализирующихся в данной области. Для этого важно избегать скучных формальных описаний, заменяя их игровым обучением в форме последовательности усложняющихся задач. Неслучайно эти игры все чаще используют в обучении [9]. Возможно, еще одна область биоинформатики, где возможен такой подход, — изучение истории хромосомных перестроек, как для кольцевых бактериальных хромосом, так и для линейных эукариотических, с различными наборами элементарных операций.
Работа выполнена при поддержке Российского научного фонда (проект 14-24-00155).
Литература
1. Rohl C. A., Strauss C. E., Misura K. M. et al. Protein structure prediction using Rosetta // Methods Enzymol. 2004. V. 383. P. 66–93. DOI: 10.1016/S0076-6879(04)83004-0.
2. Cooper S., Khatib F., Treuille A. et al. Predicting protein structures with a multiplayer online game // Nature. 2010. V. 466. P. 756–760. DOI: 10.1038/nature09304.
3. Khatib F., DiMaio F., Foldit Contenders Group et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players // Nat. Struct. Mol. Biol. 2011. V. 18. P. 1175–1177. DOI: 10.1038/nsmb.2119.
4. Gilski M., Kazmierczyk M., Krzywda S. et al. High-resolution structure of a retroviral protease folded as a monomer // Acta Crystallogr. D Biol. Crystallogr. 2011. V. 67. P. 907–914. DOI: 10.1107/S0907444911035943.
5. Khatib F., Cooper S., Tyka M. D. et al. Algorithm discovery by protein folding game players // Proc. Natl. Acad. Sci. USA. 2011. V. 108. P. 18949–18953. DOI: 10.1073/pnas.1115898108.
6. Eiben C. B., Siegel J. B., Bale J. B. et al. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players // Nat. Biotechnol. 2012. V. 30. P. 190–192. DOI: 10.1038/nbt.2109.
7. Lee J., Kladwang W., Lee M. et al. RNA design rules from a massive open laboratory // Proc. Natl. Acad. Sci. USA. 2014. V. 111. P. 2122–2127. DOI: 10.1073/pnas.1313039111.
8. Kawrykow A., Roumanis G., Kam A. et al. Phylo: A citizen science approach for improving multiple sequence alignment // PLoS One. 2012. V. 7. P. e31362. DOI: 10.1371/journal.pone.0031362.
9. Farley P. C. Using the computer game «FoldIt» to entice students to explore external representations of protein structure in a biochemistry course for nonmajors // Biochem. Mol. Biol. Educ. 2013. V. 41. P. 56–57. DOI: 10.1002/bmb.20655.

Гражданская наука
-
 01.02.2022Случайно залетевшие в Европу дальневосточные степные коньки основали там новые места зимовкиАлексей Опаев • Новости науки
01.02.2022Случайно залетевшие в Европу дальневосточные степные коньки основали там новые места зимовкиАлексей Опаев • Новости науки -
 09.06.2021Гражданская наука: приглашаются все!Валентин Власов • Библиотека • «Наука из первых рук» №5/6(90), 2020
09.06.2021Гражданская наука: приглашаются все!Валентин Власов • Библиотека • «Наука из первых рук» №5/6(90), 2020
-
 18.09.2020Наука для любителей: приглашаются все желающиеИнтервью Ольги Мустяц с Альфией Максутовой • Библиотека • «Троицкий вариант» №17(311), 2020
18.09.2020Наука для любителей: приглашаются все желающиеИнтервью Ольги Мустяц с Альфией Максутовой • Библиотека • «Троицкий вариант» №17(311), 2020
-
 13.12.2019Растительный учетАнастасия Шартогашева • Библиотека • «Популярная механика» №11, 2019
13.12.2019Растительный учетАнастасия Шартогашева • Библиотека • «Популярная механика» №11, 2019
-
 24.06.2019Результаты проекта Galaxy Zoo заставляют пересмотреть теорию образования спиральных рукавов галактикМарат Мусин • Новости науки
24.06.2019Результаты проекта Galaxy Zoo заставляют пересмотреть теорию образования спиральных рукавов галактикМарат Мусин • Новости науки
-
 19.02.2019От науки к интернет-играм: коллективное решение биоинформатических задачМихаил Гельфанд • Библиотека • «Природа» №11, 2014
19.02.2019От науки к интернет-играм: коллективное решение биоинформатических задачМихаил Гельфанд • Библиотека • «Природа» №11, 2014
-
 23.01.2019«Лагерные воришки» и гражданская наукаВероника Самоцкая • Картинки дня
23.01.2019«Лагерные воришки» и гражданская наукаВероника Самоцкая • Картинки дня
-
 07.09.2018Биологическая игра EyewireОлег Соколенко • Картинки дня
07.09.2018Биологическая игра EyewireОлег Соколенко • Картинки дня
-
 13.07.2016Полет на Луну — это командировка на неделюИнтервью Ольги Орловой с Владимиром Сурдиным • Библиотека • «Троицкий вариант» №13(207), 2016
13.07.2016Полет на Луну — это командировка на неделюИнтервью Ольги Орловой с Владимиром Сурдиным • Библиотека • «Троицкий вариант» №13(207), 2016
-
 01.01.2013Охотники за квадратамиАлександр Андреев, Олег Заикин • Библиотека • «Троицкий вариант» №24(118), 2012
01.01.2013Охотники за квадратамиАлександр Андреев, Олег Заикин • Библиотека • «Троицкий вариант» №24(118), 2012






Рис. 1. Снимок экрана компьютера с загруженной игрой Foldit [2]. Стрелками отмечено, что программа показывает пользователю в процессе его работы над пространственной структурой белка (изображенного в центре): атомы, находящиеся слишком близко друг к другу (1); водородные связи (2); открытые гидрофобные (3), гидрофильные (4) и обладающие высокой энергией (5) аминокислотные остатки. Игрок может вносить модификации в структуру, например, вводя связи (6), ограничивающие применение каких-либо операций, или «замораживая» степени свободы (7), что препятствует их изменению. Графический интерфейс пользователя также показывает текущие баллы игрока (8), рекорды других участников (9), панель с доступными операциями (10), средство общения с другими игроками (11) и инструменты создания новых «рецептов» (12)