Ферроптоз
Наталья Резник,
кандидат биологических наук
«Химия и жизнь» №10, 2018
Несколько лет назад специалисты Колумбийского университета (США) проверяли действие малых молекул на опухолевые клетки. В ходе обычных фармакологических исследований ученые открыли новый тип клеточной смерти — ферроптоз. А когда научатся им управлять, надеются обрести лекарства от рака и многих других тяжелых заболеваний.
Полочки в чуланчике смерти

Биологи привыкли все явления описывать и классифицировать. Не избегла общей участи и клеточная смерть. Ее подразделяют на случайную и регулируемую. Случайная смерть мгновенна и катастрофична: клетка гибнет от высокого давления, температуры или резкого скачка кислотности, лопается в неподходящей среде, ее, наконец, могут просто раздавить. Регулируемая смерть называется так потому, что ею можно управлять, действуя на клетку фармакологическими препаратами или влияя на работу ее генов. Если, конечно, знать молекулярные механизмы процесса и не ошибиться, потому что перечень возможных способов клеточной кончины неуклонно растет. Если до начала нынешнего века любую регулируемую гибель называли апоптозом, то теперь номенклатурный комитет по клеточной смерти предлагает нам длинный список (Cell Death & Differentiation, 2018, 25(3), p. 486–541). Клеточная танатология не стоит на месте.
Выделяют три основных типа регулируемой клеточной гибели, хорошо различимые по внешним признакам (рис. 1). Прежде всего, это апоптоз. Цитоплазма обреченной клетки сжимается, ее хроматин конденсируется, а ядро разделяется на фрагменты. В результате клетка распадается на упакованные в мембрану пузырьки — апоптозные тельца, поедаемые соседними фагоцитами. При аутофагии, то есть самоедстве, клетка сама себя переваривает в вакуолях, которых в цитоплазме образуется великое множество. Фагоциты подбирают остатки. И наконец, есть некроз, который, в отличие от апоптоза, не затрагивает клеточное ядро. Оно остается целым, зато ломается на кусочки клеточная мембрана. Мертвая клетка исчезает без видимого участия фагоцитов.
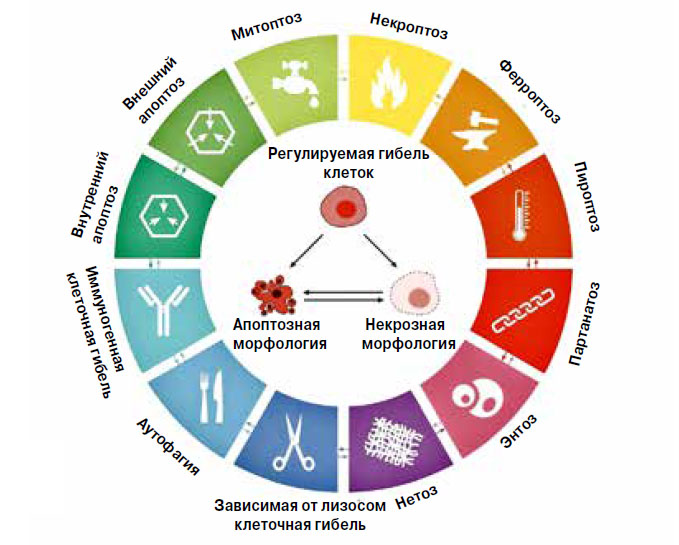
Внутри этого деления существует более тонкое, основанное на молекулярных механизмах, которые обеспечивают клеточную гибель. Классификация учитывает индукторы, рецепторы, образование новых внутриклеточных структур или изменение существующих, ферментативные и неферментативные реакции, убивающие клетку. Так, апоптоз можно разделить на внешний и внутренний, в зависимости от сигнальных путей активации: через рецепторы внешних мембран или митохондрии внутри клетки. Основные ферменты апоптоза — каспазы.
Некроз происходит без каспаз и также имеет несколько разновидностей. При митоптозе клетка умирает из-за гибели митохондрий. В результате изменений, происходящих внутри клетки, например при окислительном стрессе или избытке ионов кальция, мембрана митохондрий становится проницаемой для небольших молекул и расщепляется.
При пироптозе в плазматической мембране образуются поры, в состав которых входят определенные белки. При одной из форм пироптоза повышается температура тела, отчего этот тип клеточной смерти и получил свое название.
Зависимая от лизосом клеточная гибель — это прободение лизосомной мембраны в результате нарушения внутриклеточного гомеостаза. В лизосомах происходит расщепление макромолекул, и среда внутри них кислая. Когда она изливается в цитоплазму, клетку ничего хорошего не ждет.
Нетоз связан с внеклеточной нейтрофильной ловушкой (neutrophil extracellular trap, NET). Нейтрофилы формируют и выбрасывают во внеклеточное пространство сеть из белков и ДНК, в которую должны ловиться патогены. Однако в нее могут угодить и соседние клетки.
При энтозе одна эпителиальная клетка поглощает и поедает другую, причем в этом непременно участвует актомиозиновый комплекс. Некроптоз — реакция на изменения вне- или внутриклеточной среды, которая зависит от активации определенных рецепторов и белков внутри клетки. Для партанатоза характерны специфические повреждения ДНК в ответ на окислительный стресс, гипоксию, гипогликемию или факторы воспаления. При этом активируется фермент поли(АДФ)рибозополимераза 1 (PARP1).
Это лишь самая общая и краткая характеристика типов регулируемой гибели клеток. А последнюю карту в этом пасьянсе — ферроптоз — мы обсудим подробнее.
Железная смерть
Ферроптоз — одна из форм некроза, при которой в клетке накапливаются продукты перекисного окисления фосфолипидов, одного из основных компонентов всех клеточных мембран. Окисление происходит в присутствии ионов железа, поэтому данный тип клеточной гибели и называется ферроптозом, термин произошел от греческого слова ptosis, означающего «падать», и латинского ferrum — «железо». Его предложили первооткрыватели — профессор Колумбийского университета Брент Стоквелл и его коллеги (Cell, 2012, 149(5), p. 1060–1072).
Помимо непременного участия железа, ферроптозу свойственна еще одна особенность. Его вызывают исключительно перекиси. Ионы кислорода или свободные радикалы тоже могут окислять фосфолипиды, но эти реакции к ферроптозу не приводят. Перекисное окисление липидов присуще и другим типам клеточной смерти, в том числе и апоптозу, однако апоптоз не требует присутствия солей железа, а апоптозные ферменты не работают при ферроптозе. В нем участвуют другие ферменты, преимущественно липоксигеназы (LOX). У человека шесть вариантов LOX, у мыши — семь.
Клетки, погибающие от ферроптоза, отличить от апоптозных просто: у них целое ядро, зато митохондрии усыхают и плохо видны, как у фибробласта на фотографии в начале статьи.
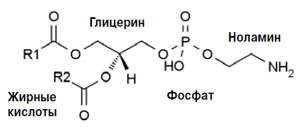
Рис. 2. Фосфатидилэтаноламин (кефалин). R1 и R2 — остатки жирных кислот
Фосфолипиды легко окисляются благодаря своей структуре. Они состоят из двух остатков жирных кислот, одна из которых, как правило, насыщенная, а другая — ненасыщенная. Эти кислоты образуют эфир с двумя гидроксильными группами глицерина. Третья гидроксильная группа образует сложноэфирную связь с фосфорной кислотой. Фосфорная кислота связана с другим спиртом, составляющим полярную головку фосфолипида (рис. 2).
На рисунке не случайно показан фосфатидилэтаноламин, именно этот тип фосфолипидов чаще всего подвергается перекисному окислению, потому что, как правило, содержит полиненасыщенные арахидоновую (C20H32O2) или адреновую (C22H36O2) кислоты. Их включение в состав фосфолипидов зависит от ферментов ACSL4 и LPCAT3 (см. таблицу).
Таблица. Ключевые ферменты ферроптоза
| Ген | Название | Функция |
|---|---|---|
| ACSL4 | Ацил-КоА синтетаза 4 | Участвует в биосинтезе фосфолипидов |
| LPCAT3 | Лизофосфатидилхолинацилтрансфераза 3 | Участвует в биосинтезе фосфолипидов |
| ALOXs | Липоксигеназы арахидоновой кислоты | Катализируют перекисное окисление арахидоновой кислоты |
| GPX4 | Глутатионпероксидаза 4 | Восстанавливает гидроперекиси липидов, подавляя ферроптоз |
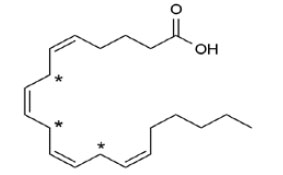
Рис. 3. Арахидоновая кислота. Чувствительные к окислению метиленовые группы между двойными связями отмечены звездочками
Двойные связи в жирных кислотах не бывают сопряженными, между ними всегда находится метиленовая группа, чрезвычайно чувствительная к атаке липоксигеназ и окислителей. В арахидоновой и адреновой кислотах по четыре двойных связи и по три метиленовых группы, что делает их особенно уязвимыми в нашем кислородном мире (рис. 3).
Перекисное окисление происходит под действием свободных радикалов, которые образуются в дыхательной цепи митохондрий или в результате других реакций. Сначала радикал атакует одну из метиленовых групп арахидоновой кислоты, отсоединяя от нее атом водорода. К этой группе арахидонатлипоксигеназа (ALOX) присоединяет молекулярный кислород, и образуется пероксильный радикал ROO•. Он может образовать новые радикалы жирных кислот, дав начало цепной реакции, либо реагирует с другой липидной молекулой и восстанавливается до гидроперекиси жирной кислоты ROOH (рис. 4).
Реакция перекисного окисления идет гораздо быстрее в присутствии ионов двухвалентного железа, которых в клетке обычно более чем достаточно. В результате реакции Фентона образуются радикалы:
Fe2+ + ROOH → Fe3+ + RO• + OH−,
Fe3+ + ROOH → Fe2+ + ROO• + H+.
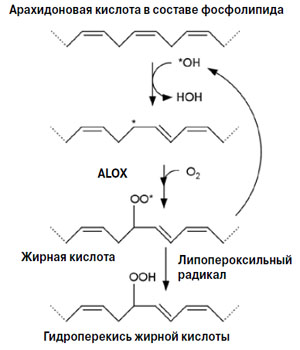
Рис. 4. Окисление полиненасыщенной жирной кислоты в составе фосфолипида
Любая реактивная молекула, способная присоединить атом водорода от полиненасыщенных жирных кислот, будь то продукты реакции Фентона или гидропероксид липидной молекулы, вызывает самоокисление липидов — это автокаталитический процесс, от липоксигеназ не зависящий, поскольку радикалы вступают в цепную реакцию.
При таком окислительном потенциале не уцелела бы ни одна фосфолипидная мембрана, не будь у клетки системы антиоксидантной защиты. В ней два основных участника. Прежде всего, это фермент глутатионпероксидаза 4 (GPX4), который восстанавливает потенциально опасные гидроперикиси липидов (LOOH) в нетоксичные спирты (LOH). Дефицит GPX4 несовместим с жизнью. Мыши, у которых этот ген нокаутирован, погибают на эмбриональной стадии. При ферроптозе активность GPX4 снижена.
Второй компонент — сам глутатион, субстрат GPX4, внутриклеточный антиоксидант. Это трипептид, состоящий из остатков глутамата, цистеина и глицина. Истощение запасов глутатиона усиливает перекисное окисление липидов и убивает клетку (рис. 5).

Рис. 5. Пути ферроптоза. ПНЖК — полиненасыщенные жирные кислоты; ФЛ-ПНЖК — фосфолипиды, содержащие полиненасыщенные жирные кислоты
Подавить!
Какие функции выполняет ферроптоз в здоровом организме, пока непонятно. Возможно, он участвует в процессе нормального эмбрионального развития конечности млекопитающих. У эмбриона лапка перепончатая, потом перепонки рассасываются, и в это время экспрессия Gpx4 понижена и замечены маркеры ферроптоза.
Зато есть много примеров связи ферроптоза с разными патологиями. Именно он повинен в повреждении тканей мозга, сердца, печени и почечных канальцев, пострадавших от ишемии / реперфузии, то есть временного кислородного голодания.
Ферроптоз дает о себе знать при обилии железа, например, в эритроцитах. Когда мышам переливали кровь с эритроцитами, поврежденными от долгого хранения, у них развивалось воспаление. Макрофаги поедали поврежденные эритроциты и погибали от ферроптоза. Он также связан с гемохроматозом печени. Это наследственное заболевание, при котором организм усваивает слишком много железа. Его излишки откладываются в разных тканях, в том числе в печени, и могут вызывать цирроз.
Ферроптоз нарушает нормальный иммунный ответ, убивая Т-лимфоциты (доказано на мышах). А еще он сопутствует нейродегенеративным заболеваниям, что неудивительно, поскольку нервные клетки отличаются максимальным содержанием полиненасыщенных жирных кислот, а некоторые патологии нервной системы, в том числе болезни Альцгеймера, Паркинсона и Хантингтона и атаксия Фридрейха вызваны неспособностью восстанавливать окисленные липиды. А еще у таких больных повышена концентрация глутамата.
В нервной системе глутамат выполняет функции нейротрансмиттера, но он же участвует в транспорте цистина. Взглянем еще раз на рис. 5. Цистин необходим для синтеза внутриклеточного антиоксиданта глутатиона. В клетку он попадает через антипортер цистин / глутамат. Клетка обменивает цистин на глутамат в соотношении 1:1. Сколько глутамата покинет клетку, столько цистина сможет войти. При избытке глутамата снаружи он из клетки не выходит, цистин внутрь не попадает, его запасы истощаются, глутатион не синтезируется, GPX4 не работает, клетка умирает. Возможно, известная токсичность глутамата для нейронов вызвана именно ферроптозом.
А еще при нейроденегеративных заболеваниях в мозге увеличивается содержание железа. Оно еще и с возрастом увеличивается, что приводит к возрастному риску ферроптоза. И многие нейродегенеративные заболевания тоже возрастные.
Получается, что ферроптоз — причина многих болезней, и его ингибиторы могли бы улучшить результаты переливания крови, защитить иммунную систему, а главное — справиться со многими тяжелыми заболеваниями, в том числе нейродегенеративными, если отыскать молекулы, способные эффективно преодолевать гематоэнцефалический барьер. Поисками лекарств — ингибиторов ферроптоза заняты многие исследователи, в том числе специалисты Мюнхенского центра им. Гельмгольца Хосе Педро Фридман Анджели и Маркус Конрад (Trends in Pharmacological Sciences, 2017, 38(5), p. 489–498).
Первыми в списке возможных антиферроптозных молекул стоят ингибиторы перекисного окисления липидов. Их можно разделить на две большие группы: ингибиторы окисления, то есть ловцы свободных радикалов, и ингибиторы липоксигеназ (рис. 6).
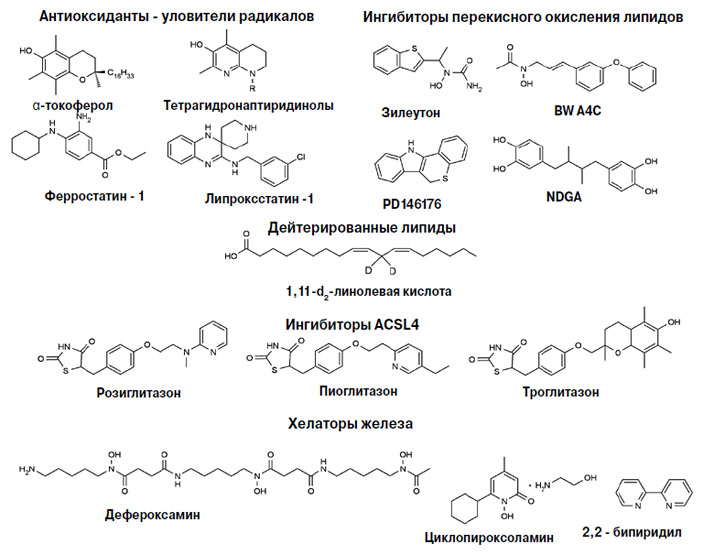
Рис. 6. Ингибиторы ферроптоза
Антиоксиданты связывают радикалы, прерывая цепную реакцию окисления. Считается, что людям полезнее всего α-токоферол, самая биологически активная форма витамина Е. Еще в 1970-х годах исследователи обнаружили, что фибробласты человека, выращенные на среде без цистина, погибают из-за нехватки глутатиона, но эту смерть может предотвратить α-токоферол. Только они не знали, что такая смерть называется ферроптозом. Антиоксиданты, в том числе и токоферол, казались многообещающим лекарством от самых разных болезней, однако не оправдали возлагаемых на них надежд (см. «Химию и жизнь» № 4, 2018). Немецкие исследователи, однако, считают, что мы еще мало знаем об антиоксидантах, во всяком случае, дефицит витамина Е связан с предрасположенностью к нейродегенеративным заболеваниям, и надо этот дефицит восполнять.
Лучше всего витамин Е проявляет себя в клеточных культурах. В присутствии α-токоферола некоторые типы клеток могут выживать даже без гена Gpx4; клетки эпителия почечных протоков и нейроны на это не способны, к сожалению. Добавки витамина Е в рацион мышей с нокаутированным геном Gpx4 восстанавливали антивирусный и антипаразитарный ответ Т-клеток и прекращали дегенерацию гепатоцитов. Правда, все эксперименты на мышах проводят при концентрации витамина Е, по крайней мере в два раза превышающей физиологическую.
Исследователи разрабатывают синтетические аналоги α-токоферола, возлагая особые надежды на тетрагидронаптиридинолы. Эти стабильные соединения, защищенные от самоокисления, в растворах и липосомах реагируют с радикалами почти в 30 раз быстрее α-токоферола.
Антиоксиданты липроксстатины и ферростатины обнаружили в результате целенаправленного скрининга соединений, способных ингибировать ферроптоз. Первый найденный липоксстатин, Lip-1, подавляет ферроптоз в наномолярной концентрации, молекула улучшает состояние почек у мышей с нокаутированным геном Gpx4. Потеря этого гена вызывает острую почечную недостаточность. Ферростатин Fer-1 в пробирке защищает липидный бислой от окисления лучше, чем α-токоферол. Оба соединения предохраняют ткани печени и почек от последствий ишемии / реперфузии. Механизмы их действия сейчас исследуют.
О работе ингибиторов липоксигеназ известно больше, однако ни одну липоксигеназу не удалось убедительно связать с ферроптозом. А еще сложность в том, что этот фермент имеет много изоформ и каждая требует специфического ингибитора.
Липиды, у которых в метиленовой группе атомы водорода заменены на атомы дейтерия, представляют собой гораздо более слабый субстрат для липоксигеназ. Клетки, растущие в питательной среде с дейтерированной линолевой кислотой, оказались защищенными от ферроптоза в клеточных моделях неврологических заболеваний. Однако неясно, насколько это средство безопасно и эффективно in vivo, то есть можно ли использовать его как лекарство.
Поскольку при ферроптозе прежде всего окисляются арахидоновая и адреновая кислоты, можно защитить фосфолипиды от окисления, предотвратив встраивание этих кислот в молекулу. Для этого используют ингибиторы ферментов ACSL4 и LPCAT3. Клетки с дефицитом ACSL4 устойчивы к ферроптозу. Во всяком случае, мыши, которым давали розиглитазон, реже умирали от острой почечной недостаточности, вызванной нокаутом Gsp4.
И наконец, ферроптоз предотвращают хелаторы железа — вещества, образующие с ним химический комплекс.
Использовать!
Было бы ошибкой думать, что с ферроптозом всегда нужно бороться. Считается, что он подавляет рост опухолевых клеток, и это одна из функций, выполняемых им в организме. И обнаружили ферроптоз в процессе поиска лекарства от рака. Если ферроптоз действительно убивает некоторые раковые клетки, а эксперименты, поставленные на клеточных культурах и мышах, дают основание на это надеяться, вещества, стимулирующие ферроптоз, могут стать лекарством.
Брент Стоквелл и его коллеги делят индукторы ферроптоза на четыре основные группы (Cell, 2017, 171(2), p. 273–285). Прежде всего, это вещества, блокирующие транспорт цистина в клетку (рис. 7).

Рис. 7. Индукторы ферроптоза
Для работы GPX4 необходим глутатион, а для синтеза глутатиона — цистеин. Если заблокировать транспорт цистина в клетку, запасы глутатиона истощатся, GPX4 утратит активность и клетка погибнет. Однако некоторые клетки могут использовать для биосинтеза цистеина метионин, и они устойчивы к ферроптозу. Ученые обнаружили несколько молекул, блокирующих транспорт цистина (эрастин и имидазолкетонэрастин, сорафениб, сульфасалазин).
Вторая группа индукторов ферроптоза — непосредственные ингибиторы GPX4. Они взаимодействуют с молекулой, подавляя ее ферментативную активность. Известно несколько таких соединений, в том числе RSL3 (RASselective lethal 3).
Эрастин и RSL3 убивают 177 линий опухолевых клеток и ограничивают рост опухолей, привитых мышам. К сожалению, они малорастворимы в воде и нестабильны, поэтому годятся только для работы с клеточными культурами. В качестве лекарства их использовать нельзя. Зато имидазолкетонэрастин, сорафениб и некоторые другие молекулы — можно.
Есть соединения, которые истощают запасы GPX4 и липофильного внутриклеточного антиоксиданта CoQ10. В их числе FIN56 (индуктор ферроптоза 56) и CIL56 (каспаза-независимая леталь 56). CIL56 может активировать и другие типы клеточного некроза, а FIN56 — только ферроптоз.
Для активации ферроптоза можно использовать индукторы перекисного окисления, например FINO2 (индуктор ферроптоза эндопероксид). Он стимулирует перекисное окисление липидов и окисляет железо.
И наконец, есть соединения, которые вызывают ферроптоз в определенных тканях: четыреххлористый углерод в печени, артесунат — в поджелудочной железе, цисплатин и производные артемизинина вызывают в некоторых тканях и ферроптоз, и апоптоз.
Сейчас такое время, что любую молекулу, любое соединение объявляют лекарством от рака или, по крайней мере, подозревают в таких способностях. Иначе как-то даже неприлично. Но индукторы ферроптоза действительно позволяют надеяться. Некоторые особо злокачественные клетки, уцелевшие после химиотерапии, оказались очень чувствительны к ингибиторам GPX4 и вообще к ферроптозу.
Где спрятан ферроптоз?
Перекисное окисление липидов и влияние железа на состояние клетки известны давно. Возможно, и ферроптоз ученые описывали неоднократно, прежде чем разобрались в его молекулярном механизме, выделили в отдельный тип клеточной смерти и придумали ему название. Понимание пришло в 2012 году, и с тех пор исследования ферроптоза идут семимильными шагами. Тем не менее пока трудно сказать, почему эволюция заложила в клетку такую мину — ведь железо и кислород у нас повсюду. Возможно, дело в том, что включение фосфолипидов в состав мембраны делает ее прочной и пластичной. Такая мембрана дает клеткам возможность формировать сложные нейронные сети и существовать в средах с разными температурами. Преимущества фосфолипидной мембраны столь велики, что перевешивают ее восприимчивость к перекисному окислению. Оно может происходить в мембранах любых органелл: митохондрий, лизосом, эндоплазматического ретикулума. Чувствительность органелл к ферроптозу различна и зависит от состава липидов, запасов железа, количества глутатиона, уровня экспрессии LOXs и локализации GPX4. Но, что интересно, механизм убийственного действия ферроптоза непонятен до сих пор (PLoS Biology, 2018, 16(5), e2006203).
Есть, конечно, разные гипотезы. Возможно, окисленные фосфолипиды могут переориентироваться и переходить в водную фазу, что приводит к истончению мембраны и привлекает к клеткам макрофагов. Действительно, во время ферроптоза мембрана становится более доступной для окислителей и образует мицеллы. Согласно другой гипотезе, окисленная мембрана становится более пористой и меняет проницаемость. Тем не менее перекисное окисление мембран митохондрий или лизосом не приводит к ферроптозу. Эксперименты с клетками, лишенными митохондрий, показали, что митохондрии для ферроптоза не требуются. Исследователи предположили, что ферроптоз таится в мембранах эндоплазматического ретикулума (ЭР). Это структура, в которой происходит белковый синтез, а потом белковые молекулы укладываются в правильную трехмерную конфигурацию и выходят в цитоплазму. Если белки не могут правильно сложиться, они остаются в ЭР и забивают его — такая ситуация называется эндоплазматическим стрессом. Он приводит ко многим патофизиологическим нарушениям, включая дефицит глюкозы, гипоксию, неправильную регуляцию концентрации ионов кальция и вирусную инфекцию. Если блокировать доступ цистина в клетку, в ней развиваются ЭР-стресс и ферроптоз, так что между этими явлениями может быть связь.
Эту гипотезу сейчас активно проверяют, и когда точно установят роль ферроптоза в организме, механизм его убийственного действия и возможные средства воздействия, то с радостью помогут больным злокачественными опухолями, пациентам с болезнью Альцгеймера и жертвам ишемии.











Рис. 1. Типы регулируемой гибели клеток