О молекулярных механизмах возникновения опухолей
Федор Киселев
«Природа» №4, 2014
 Об автореФедор Львович Киселев — член-корреспондент РАМН, профессор, доктор биологических наук, руководитель отдела трансформирующих генов опухолей НИИ канцерогенеза Онкологического научного центра им. Н. Н. Блохина. Занимается изучением молекулярных механизмов канцерогенеза. |
Первое десятилетие XXI в. принесло ошеломляющие данные о механизмах возникновения опухолей и изменили наши представления об этих болезнях. Какие подходы к диагностике и терапии открыли новые знания? Попытаюсь рассказать обо всем этом.
Немного истории
Возникновение молекулярной онкологии во второй половине прошлого века было связано с обнаружением двух типов генов: стимулирующих клеточный рост (онкогенов), и, наоборот, ограничивающих его (генов-супрессоров) [1]. Как тогда предполагали, именно взаимодействие этих генов служит основной причиной неконтролируемого роста клеток, т. е. появление опухоли. Такие революционные для онкологии факты имеют свою историю.
Огромную роль в развитии молекулярной онкологии, на мой взгляд, сыграла вирусная теория происхождения опухолей, высказанная Л. А. Зильбером1 в середине 40-х годов и спустя 20 лет получившая всеобщее признание как вирусогенетическая теория происхождения опухолей [2, 3]. Ее основные положения гласили:
- все опухоли человека вызываются вирусами;
- вирус лишь инициирует перерождение нормальной клетки в опухолевую;
- вирусная нуклеиновая кислота встраивается в геном клетки, модифицируя ее генетическую программу так, что клетка становится бессмертной.
Первый и единственный вирус у человека выделили в конце 60-х годов у детей, страдающих одной из форм онкологической болезни в Африке, — это был вирус лимфомы Беркитта. В 70–80-х годах вирусная теория продолжала свое триумфальное шествие — было обнаружено большое количество РНК- и ДНК-вирусов, вызывающих опухоли у самых различных видов млекопитающих и птиц.
Изучение РНК-содержащих онкогенных вирусов позволило сделать новый, принципиальный шаг в понимании механизмов злокачественного роста. У птиц эти вирусы делятся на две основные группы: вызывающие либо лейкозы, либо саркомы.
Оказалось, что РНК вируса куриной саркомы Рауса, приводящего к развитию опухоли у птиц и к злокачественному перерождению (трансформации) культивируемых клеток, содержит фрагмент, который отсутствует в РНК лейкозных вирусов. Выяснилось, что именно он отвечает за трансформацию клеток, способных к неограниченному размножению в культуре клеток, а при введении чувствительным животным вызывает у них опухоли. Фрагменты генома со сходными свойствами обнаружили во всех РНК-содержащих вирусах, инициирующих опухоли, и назвали их онкогенами. За эти работы, выполненные в конце 70-х годов, М. Бишоп и Х. Вармус в1989 г. были удостоены Нобелевской премии2.
Изучение генома опухолевых клеток, зараженных саркомными вирусами, принесло новую сенсацию — гомологи вирусных онкогенов есть во всех нормальных клетках. Естественно возник вопрос: почему же в них не проявляется онкогенный потенциал? И проявляется ли он вообще? И если да, то как?
Гены-активаторы и гены-супрессоры
Изучение структуры клеточных онкогенов (протоонкогенов), их локализации на хромосомах, роли в жизни клетки и возможных механизмов активации показало, что протоонкогены действительно могут осуществлять контроль за делением клеток [1–3]. В качестве индукторов клеточного размножения молчащие протонкогены способны активироваться в результате следующих генетических процессов:
- точечных мутаций в кодирующих последовательностях онкогена, что изменяет структуру его белкового продукта, соответственно и взаимодействие белка с мишенью;
- перемещения отдельных фрагментов между хромосомами (транслокации);
- амплификации гена (увеличение его копийности);
- усиления работы некоторых онкогенов во многих опухолях, т. е. повышенный синтез белкового продукта.
Важным шагом стало также выяснение роли онкобелков при прохождении сигнала к делению (активной пролиферации) от клеточной поверхности в ядро. В результате были выделены четыре группы таких белков. В первую входили внеклеточные ростовые факторы; во вторую — продукты онкогенов, служащие рецепторами для этих факторов (рецепторные тирозинкиназы); в третью — онкобелки (кодируемые онкогенами семейства RAS и большая группа серин-треониновых киназ), осуществляющие передачу ростового сигнала в ядро; в четвертую — продукты ядерных онкогенов (MYC, JUN, MYB), способные контролировать синтез ДНК. Особенно интересны киназы, которые фосфорилируют белки-мишени по тирозину, также по двум другим аминокислотам — серину и треонину.
Итак, к концу прошлого века стало ясно, что в геноме нормальных клеток имеется достаточно большое количество генов — потенциальных индукторов опухолевого роста. Они функционируют на разных этапах передачи сигнала к размножению клеток, а их активация может приводить к бесконтрольному клеточному делению, т. е. к образованию опухоли.
Другая группа генов — это супрессоры опухолевого роста [4]. Первый из них — ген ретинобластомы (Rb). Его существование постулировал в 1971 г. А. Кнудсон, изучая спорадические и наследственные формы опухолей сетчатки глаза (ретинобластомы) [5]. Цитогенетическими исследованиями на хромосоме 13 была обнаружена небольшая делеция, в которой затем идентифицировали ген Rb (он был либо утерян, либо инактивирован в ретинобластомах). Позднее ген Rb клонировали. Он обладает супрессорным эффектом на трансформированные клетки и работает во всех нормальных клетках, в которых его продукт (pRb105) обратимо фосфорилирован. В неделящихся клетках он теряет остаток фосфорной кислоты и образует комплексы с белками (гистоновыми деацетилазами — HDAC), вызывающими изменения хроматина, и с транскрипционными факторами семейства Е2F.

Эти факторы — основные регуляторы синтеза белков, контролирующих нормальный клеточный цикл. Транскрипция их генов подавлена, когда фактор Е2F находится в комплексе с Rb/HDAC. При митогенном сигнале в начальной фазе (G1) клеточного цикла белок Rb фосфорилируется одной из циклин-зависимых киназ (ферментов, регулирующих клеточный цикл), а фактор E2F, который активирует многие гены, в том числе и необходимые для синтеза ДНК, освобождается. После перехода клетки в S-фазу белок Rb теряет остатки фосфорной кислоты (дефосфорилируется). Кроме клеточного цикла ген Rb через свой продукт участвует в дифференцировке клеток, репарации ДНК и ее репликации, а также в регуляции апоптоза. Таким образом, ген Rb, модулируя через свой белок активность транскрипционного фактора E2F и регулируемых им генов, играет ключевую роль в контроле последовательных событий, обеспечивающих жизненный цикл клетки. Значительная часть мутаций гена Rb (кроме ретинобластомы они обнаружены и в других опухолях) приводит либо к потере его функций, либо к прекращению синтеза РНК, либо к образованию белка с измененной структурой. Транскрипционный фактор E2F при этом находится в перманентно активированном состоянии. В итоге негативная регуляция клеточной пролиферации прекращается.
Еще один широко распространенный ген-супрессор опухолевого роста кодирует белок с молекулярной массой 53 кДа, отсюда и его название — p53 [4]. В опухолях человека этот ген наиболее подвержен мутациям — они обнаруживаются более чем в 50% случаев и распределены по всему гену. В нормальных, неделящихся клетках p53 обладает очень слабой транскрипционной активностью. Модификации, происходящие при стрессах или внутриклеточных повреждениях, которые изменяют структуру белка, существенно увеличивают транскрипционный потенциал p53. Основными мишенями при этом служат белки, контролирующие апоптоз и клеточный цикл. Следовательно, р53 в случае каких-либо серьезных воздействий на ДНК отвечает за «охрану» клеток: либо индуцирует репарацию повреждений, либо останавливает клеточный цикл, либо стимулирует клетки с измененным геномом к апоптозу.
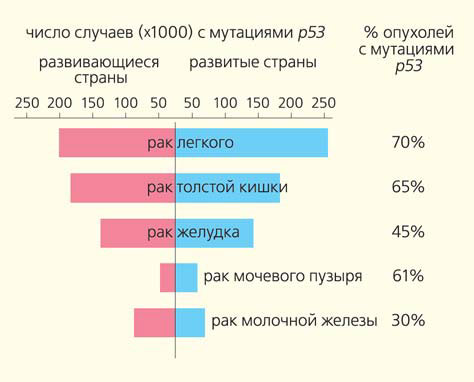
Мутации в гене р53— наиболее часто встречаемое изменение в опухолях. Прямоугольники — частота определенного рака на 250 тыс. населения; числа справа — количество (%) опухолей с мутациями р53 в данном типе рака
В настоящее время имеется более 100 генов-мишеней для транскрипционной активности р53. Среди них можно выделить несколько функционально различных групп: контролирующие через свои продукты апоптоз; ангиогенез (формирование новых сосудов в опухоли); регулирующие клеточный цикл (это циклины, циклин-зависимые киназы и их ингибиторы); морфологию и/или миграцию клеток. Одна из важных функций р53 — репрессия гена каталитической субъединицы теломеразы, фермента, важного в репликативном старении клеток. Следовательно, изменение активности гена р53 обеспечивает появление характерных для неопластических клеток свойств, которые за короткий срок меняют их генетическую программу. Это объясняет тот факт, что в опухолях различных типов мутации в р53 встречаются чаще, чем в других генах. Упомянутые гены-супрессоры не единственные, к настоящему времени их обнаружено больше 10 и с каждым годом их количество возрастает, причем их функциональная активность специфична для разных опухолей.
Исследования генов, которые через кодируемые ими белки контролируют пролиферацию клеток и играют главную роль в их бесконтрольном делении, проведенные в основном в конце прошлого столетия, убедительно показали, что рак — заболевание генетического аппарата клеток. Следовательно, точные знания обо всех генетических составляющих канцерогенеза и сравнения нуклеотидных последовательностей ДНК в опухолевых и нормальных клетках совершенно необходимы. Такие работы стали возможными после того, как геном человека был полностью расшифрован.
Специфические свойства опухолей и молекулярные механизмы рака
Однако прежде чем перейти к рассмотрению этих данных, вернемся к началу статьи — к вопросу о роли вирусов в возникновении опухолей человека.
Если в 1960-х годах был обнаружен только вирус лимфомы Беркитта, принадлежащий к группе герпесов и приводящий к лимфоме у африканских детей, то к концу века стали известны еще несколько вирусов, связанных с опухолями человека. Это ДНК-содержащие вирусы: герпеса тип 8 (или саркомы Капоши), вызывающий один из видов саркомы кожи; гепатита В, с высокой частотой выявляемый в опухолях печени; нескольких типов папиллом человека (этиологический агент при раке шейки матки) и саркомы Меркеля3, принадлежащий к вирусам группы Papova (вирусы полиомы и SV40, которые вызывают опухоли у мышей и обезьян). С опухолями человека ассоциированы также два РНК-содержащих вируса — гепатита С в опухолях печени (механизм проявления его онкогенного потенциала еще точно не установлен) и T-клеточного лейкоза взрослых, связанный только с одной и редкой формой лейкоза в нескольких эндемичных районах мира. РНК этого вируса сходна по своей структуре с другими лейкозными вирусами млекопитающих и птиц.
К настоящему времени установлены основополагающие принципы, по которым вирусы изменяют генетическую программу клеток, модифицируя ее таким образом, что клетка теряет контроль над собственным делением. ДНК-вирусы содержат гены, белковые продукты которых инактивируют гены-супрессоры опухолевого роста, а в РНК-вирусах имеются гены, опосредованно ускоряющие процессы клеточной пролиферации [6, 7].
Из всех опухолей человека по частоте встречаемости около 20% ассоциированы с вирусами. Большим достижением в борьбе с раком стало создание поливалентной вакцины против опухолей шейки матки, вызванных вирусами папилломы. Применение вакцины практически полностью исключает появление предопухолевых поражений и опухолей4.

Опухоли человека, возникновение которых связано с вирусами. На оси абсцисс — типы опухолей, возникновение которых связано с вирусами. На оси ординат — показатель (%) летального исхода, вызванного определенным типом рака от общего числа смертности от всех опухолей
С наступлением XXI в. начался новый этап в понимании молекулярных механизмов опухолевого роста. Прогресс шел в двух направлениях: изучались биологические свойства опухолевых клеток, отличающие их от нормальных; и велась работа по расшифровке молекулярных механизмов, контролирующих возникновение этих специфических свойств. В 2011 г. появился обзор, в котором были суммированы признаки опухолевых клеток [8]:
- отсутствие необходимости дополнительных сигналов к делению (пролиферации);
- потеря способности реагировать на сигналы, которые сдерживают пролиферацию;
- замедление процессов программируемой клеточной гибели (апоптоза);
- неограниченный репликативный потенциал (преодоление так называемого лимита Хайфлика — не более 30–50 циклов репликации);
- инвазия и метастазирование (распространение опухолевых клеток внутри поражаемого органа и их перенос кровью в другие органы, прежде всего в лимфоузлы) — ключевые стадии злокачественного роста;
- геномная нестабильность (ускоренное накопление мутаций);
- формирование сосудов в опухоли, без которых невозможно накопление опухолевой массы (активация ангионеза).
- адаптация окружающих опухолевый очаг стромальных компонентов к потребностям его роста;
- ускользание от иммунного надзора, связанное, возможно, с селекцией определенного клона опухолевых клеток, которые в наименьшей степени изменяют свою антигенную структуру;
- воспаление, почти всегда предшествующее и/или сопутствующее опухолевому росту и создающее благоприятную среду для увеличения клеточной массы;
- аэробный гликолиз, который, видимо, может поддерживать злокачественный рост, способствуя более эффективному синтезу макромолекул и органелл, необходимых для новых клеток.
Расшифровка в начале XXI в. генома человека стимулировала стремительный прогресс в изучении молекулярных механизмов злокачественного роста. Появилась возможность сравнивать геномы нормальной и опухолевой клеток, т. е. их полную нуклеотидную последовательность ДНК [9]. Такие работы проводились огромными научными интернациональными коллективами, которые включали десятки лабораторий и сотни исследователей. В результате оказалось, что в геноме опухолевых клеток мутации возникают в десятки раз чаще, чем в нормальных клетках. В большинстве своем эти мутации не влияют на онкогенный потенциал клетки, поэтому их назвали пассажирами. В запуске злокачественного преобразования клеток участвует лишь небольшая часть соматических мутаций (их называют водителями; по-английски driver). Именно они особенно интересны как потенциальные мишени для противоопухолевой терапии.
Определен также широкий спектр генетических повреждений в опухолях — от точковых мутаций до микроделеций (потери нескольких нуклеотидных пар) или микроинсерций (включения дополнительных пар нуклеотидов). Их последствия, как правило, сводятся к изменению структуры кодируемого мутантным геном белка, в результате чего он теряет способность взаимодействовать с мишенью. В ряде случаев нуклеотидные замены могут приводить к образованию стоп-кодона, т. е. к остановке синтеза полноразмерного белка и, значит, к потере его природных функций. Фенотипические проявления микроделеций и микроинсерций напрямую зависят от количества вовлеченных нуклеотидов. Если их число кратно трем, то рамка считывания и функции белка сохраняются (три нуклеотида могут кодировать одну аминокислоту). Так активируется рецептор эпидермального фактора роста EGFR (epidermal growth factor receptor) в опухолях легких. Однако в большинстве случаев при утрате или появлении новых нуклеотидов рамки считывания сдвигаются (при этом нарушается последовательность нуклеотидов, кодирующих нормальный белок) и тогда синтезируется неправильный белок, лишенный активности. Для большинства генов-супрессоров характерен именно такой механизм.
Многие опухоли отличаются нестабильностью протяженных участков хромосом, поэтому для них типичны нарушения копийности (амплификации) генетического материала. При ее увеличении количество белковых продуктов онкогенов возрастает. Значительно чаще амплификаций встречаются делеции участков хромосом.

Транслокация участка хромосомы 9, содержащей онкоген ABL, на хромосому 22 в район участка BCR, в результате чего образуется химерный ген ВСR/ABL. Такая химерная хромосома 9, получившая название филадельфийской, служит маркером хронического миелоидного лейкоза
Еще один пример макромутаций в опухолях — транслокации участков хромосом, которые изменяют активность генов или приводят к образованию химерных белков. Такие мутации характерны главным образом для онкогематологических патологий. Примером служит перестройка ВСR/ABL, лежащая в основе хронического миелоидного лейкоза, или транслокации тирозинкиназных генов ALT и RET, наблюдаемые в карциномах легких.
В целом, в превращении нормальной клетки в опухолевую участвует ~400 генов, несущих мутации, что составляет около 2% генов, кодирующих белки. Выявлено от 1000 до 10 тыс. возможных мутационных замещений в опухолях молочных желез, яичников, при колоректальном раке, раке поджелудочной железы и глиомах (опухолях мозговой оболочки). Существенно меньше мутаций обнаружено в опухолях мозга, яичка, острых лейкозах, а наиболее агрессивные опухоли легких и меланома содержат более 10 тыс. генетических изменений. Таким образом, множественные мутации представляют непременный атрибут опухолевой клетки, но их спектр специфичен для каждой конкретной опухоли, т. е. каждая из них имеет собственную генетическую программу.
Полная расшифровка генома человека показала, что в синтезе структурных белков, необходимых для функционирования клетки, участвует менее 10% ее генома. А каковы функции остальных 90% нуклеотидных последовательностей ДНК и как регулируется работа мутантных генов? На эти вопросы предстояло найти ответы.
Эпигенетические нарушения
Дальнейшие исследования убедительно продемонстрировали, что опухолевым клеткам присущи два типа генетических нарушений — мутационные и эпигенетические. Последние возникают при различных патологиях, включая опухоли, не связанные с нарушением первичной структуры гена. К таким эпигенетическим изменениям относят:
- метилирование ДНК (присоединение метильной группы к цитозину);
- посттрансляционные модификации гистонов (основной группы белков, формирующих хроматин);
- расположение нуклеосом на ДНК;
- образование и функционирование некодирующих РНК (микроРНК).
Остановимся на вопросах, связанных с метилированием ДНК, модификацией гистонов и функционированием микроРНК, прежде всего потому, что они могут представлять интерес для клинической практики. Метилирование цитозина в составе ДНК было открыто Б. Ф. Ванюшиным и А. Н. Белозерским еще в середине 70-х годов. Они показали, что в животных клетках происходит метилирование цитозина в паре нуклеотидов гуанин-цитозин (CpG), которые разбросаны по всему геному. Сегодня мы знаем, что в геноме человека существует множество участков, обогащенных этой парой (так называемые CpG-островки), но только их метилирование внутри промоторов (участков ДНК, с которых начинается синтез РНК) и первых экзонов гена подавляет его работу. Процесс этот наследуется клеткой, но он обратим. Такие агенты, как 5-азацитидин, отщепляют метильную группу, что приводит к активации транскрипции. В опухолевых клетках часто метилируются гены, играющие главную роль в канцерогенезе. К ним относятся гены, повреждающие ДНК и репарирующие ее; гены-супрессоры; регуляторы апоптоза, адгезии клеток; гены, участвующие в ангиогенезе, иммунном ответе; гены микроРНК.

Схема основных эпигенетических изменений в опухолевой клетке — метилирования ДНК и ацетилирования гистонов
Таким образом, метилирование генов, контролирующих основные этапы пролиферации, может оказывать тот же эффект на развитие опухолей, что и структурные мутации. В настоящее время метилирование принято обозначать термином «эпимутация», поскольку оно не изменяет нуклеотидную последовательность гена. Метилированию могут подвергаться и другие CpG-обогащенные участки внутри гена, но, как и в случае мутаций-пассажиров, принципиальных изменений в функциональной активности гена не происходит.
Важно дополнить, что метилирование может проявляться на самых ранних стадиях канцерогенеза и во всех типах опухолей, но набор метилированных генов специфичен для каждой [10].
Еще один уровень регуляции транскрипции — это модификация гистонов, ядерных белков, образующих комплекс с ДНК (хроматин). Известны четыре вида гистонов. Две молекулы каждого из них составляют нуклеосому — структуру, обвитую фрагментом ДНК длиной 146 нуклеотидных пар. В настоящее время обнаружено 16 модификаций гистонов, которые могут изменять конформацию нуклеосомы и таким образом влиять на репликацию ДНК и транскрипцию. Из возможных модификаций хорошо изучены метилирование и ацетилирование, катализируемые специфическими гистоновыми ферментами, которые осуществляют и обратный процесс. Сейчас уже получены картины распределения ацетилирования и метилирования гистонов в первичных опухолях и в культивируемых опухолевых клетках. Для последних характерна значительная потеря метилирования гистона Н4, который служит одним из маркеров неактивного хроматина [11, 12].
Другая стремительно развивающаяся область эпигеномики опухолей — изучение некодирующих РНК [13]. В ходе исследований по международной программе ENCODE (the ENCyclopedia of DNA Elements) выяснилось, что не менее 75% генома способно транскрибироваться, но более половины этих фрагментов относятся к некодирующим РНК. Они делятся на две большие группы: длинные (от 200 до 10 тыс. рибонуклеотидов) и малые (менее 200), среди которых лучше всего изучен класс микроРНК (miR). Они выполняют важнейшие регуляторные функции в жизни нормальных клеток, а нарушения в их работе обнаружены при многих заболеваниях человека, в том числе и онкологических. Подтверждено, что микроРНК не участвуют в синтезе белков и кодируются специальными генами.
МикроРНК (в настоящее время их известно более 2000) — негативный регулятор экспрессии генов. Некодирующие однонитевые молекулы микроРНК (из 18–25 рибонуклеотидов) взаимодействуют с комплементарными участками информационной РНК (иРНК), что приводит либо к их деградации, либо препятствует трансляции в белки в рибосомах. Гены микроРНК эволюционно консервативны и распределены по всему геному человека (в некоторых участках они образуют специфические кластеры). Кодирующие их участки генома расположены на разных хромосомах. Благодаря небольшому размеру каждая микроРНК, как правило, может взаимодействовать с несколькими иРНК, имеющими комплементарные участки. МикроРНК регулируют более 30% генов человека, участвующих в клеточной дифференцировке, кроветворении, устойчивости к стрессу, метаболизме, клеточной пролиферации и апоптозе. Такой широкий спектр генов, регулируемых микроРНК, свидетельствует о том, что нарушения в их работе могут существенно влиять на все стадии канцерогенеза — от возникновения опухоли до образования метастазов.

Роль микроРНК в проявлении свойств, характерных для опухолевых клеток. По кругу выделены основные биологические свойства опухолевых клеток и обозначены типы микроРНК (miR), участвующие в данном процессе
Характерная особенность опухолевых клеток — нарушения в работе микроРНК. Существует несколько механизмов дисрегуляции в опухолях: делеции, точечные мутации, геномные транслокации локуса микроРНК, т. е. изменения, присущие и структурным генам.
Каждая опухоль имеет специфический набор микроРНК. Но среди них можно выделить те, которые наиболее часто встречаются в определенных опухолях. Так, miR-121 экспрессируется в опухолях трех различных локализаций, в опухолях шести типов часто наблюдается повышенная транскрипция miR-21, а в пяти из них — miR-17-5p и miR-191. Следовательно, некоторые микроРНК могут регулировать клеточные процессы, общие для нескольких типов рака.
В зависимости от того, работу какого гена подавляют микроРНК, их функции в канцерогенезе могут быть сходными с действием либо онкогенов, либо генов-супрессоров. Те микроРНК, гены которых амплифицированы или очень активны, работают как онкогены. Как правило, в каждой опухоли наблюдается повышение экспрессии нескольких типов микроРНК и их спектр специфичен для каждой опухоли. Но существуют микроРНК, которые активируются в нескольких типах опухолей: miR-121 — в опухолях легких, молочных желез, печени и при колоректальном раке, а miR-221 — в опухолях молочных желез и простаты.
Другие микроРНК, в генах которых есть делеции или их экспрессия снижена, уподобляются опухолевым супрессорам. В модельных системах эти микроРНК могут ограничивать рост опухолевых клеток, индуцировать апоптоз в культуре клеток или при их имплантации чувствительным животным. Спектр микроРНК со сниженным уровнем экспрессии также специфичен для каждого типа опухолей, но подавления одними и теми же микроРНК могут быть выявлены в разных опухолях, к примеру miR-143 и miR-145 в опухолях молочных желез, при колоректальном раке и раке простаты.
Интересно, что для одних и тех же микроРНК в одних опухолях экспрессия повышена, а в других — наоборот, снижена. Например, экспрессия кластера mi-let 7 повышается в опухолях молочных желез и, наоборот, снижается в опухолях легких.
От того, какие именно микроРНК экспрессируются в опухолевых клетках, зависят их свойства, необходимые для поддержания трансформирующего потенциала. Например с miR-7 связана устойчивость к ингибиторам ростовых факторов; с miR-155 — уход от иммунного ответа; с miR-1296 — включение бессмертного деления; с miR-155 и miR-29а — воспаление, стимулирующее опухолевый процесс; с miR-205 — активация инвазии и метастазирования; с miR-29b — индукция ангиогенеза; с miR-521 — мутации и генетическая нестабильность; устойчивость к апоптозу зависит от четырех видов — miR-20а, -21, -24, -133а; дисрегуляция энергетических процессов — от miR-122 и -210. Активность перечисленных видов микроРНК специфична для каждой опухоли и проявляется дифференцированно в конкретной опухоли.
Спектр экспрессии микроРНК в первичной опухоли отличен от такового в метастазах: выявлены микроРНК, усиливающие метастатический потенциал клеток, и другие, наоборот, ингибирующие этот процесс. Это известно для всех изученных в настоящее время первичных опухолей и их метастазов. Спектр активирующих и ингибирующих микроРНК специфичен для каждого типа опухоли. Естественно, что мишенями для таких РНК служат разные гены. Экспрессия микроРНК в нормальных клетках специфична для каждой ткани и, видимо, может отражать происхождение опухоли и степень ее дифференцировки. Важное свойство микроРНК — их способность мигрировать в кровь. Они, в отличие от информационных РНК, очень стабильны. Это, вероятно, определяется их способностью формировать частицы, подобные эндосомам, защищающим от нуклеаз. Содержание микроРНК в крови или плазме можно оценить с помощью полимеразной цепной реакции и некоторых других методов. Такие свойства микроРНК пригодны для прогноза рака, его диагностики, а также для четкой классификации опухолей и мониторинга ответа на лечение [14].
Итак, молекулярные механизмы опухолевого роста, исследованные в первое десятилетие XXI в., открыли принципиально новые возможности ранней диагностики и терапии опухолей. Основаны эти новшества на обнаружении генетических и эпигенетических нарушений, контролирующих размножение опухолевых клеток.
Молекулярная диагностика и терапия
Как уже понятно из сказанного, в XXI в. исследования в онкологии, проводимые для целей диагностики и терапии, перешли на молекулярный уровень. Новые диагностические и терапевтические подходы принципиально отличаются тем, что в основе их применения лежит анализ генетического материала клетки. Геномные и эпигеномные нарушения стало возможным идентифицировать в образцах опухолевой ткани; парафиновых блоках срезов опухолей; в ДНК, циркулирующей в крови больных; и микроРНК, которая содержится в эндосомах крови или в самой опухоли.
Первые разработки противоопухолевых препаратов основывались на том, что опухолевая клетка активно делится, поэтому и цитотоксическая терапия была направлена на удаление быстро делящихся клеток. С появлением молекулярной онкологии акценты в лечении сместились — в центре внимания оказались отдельные биологические макромолекулы, выступающие в роли пускового механизма канцерогенеза. Возникла так называемая таргетная (англ. target — ‘цель’, ‘мишень’) терапия, направленная на определенные клеточные мишени — гены или их белковые продукты. В настоящее время оба вида терапии — цитотоксическая и таргетная — взаимно дополняют друг друга.
Какие же модификации генов и их белковых продуктов могут быть полезны для поиска таргетных препаратов, что может быть мишенью в этом случае?
Открытие онкогенов послужило спусковым механизмом для этих поисков. Изучение белковых продуктом онкогенов показало, что значительное число активированных молекул относится к классу протеинкиназ — ферментов, участвующих в передаче внутриклеточных сигналов посредством фосфорилирования белков-мишеней.
Разработка специфических ингибиторов киназ оказалась чрезвычайно эффективной в поиске средств лечения рака. Сегодня именно антагонисты фосфорилирования представляют собой самый большой класс таргетных препаратов.
Наиболее известная мишень — рецептор эпидермального фактора роста EGFRB-2, или HER2. Его ген кодирует тирозинкиназу, которая способна самостоятельно передавать сигналы от клеточной мембраны к ядру. Этот ген активируется в опухолях молочных желез, и первый таргетный препарат — антитело герцептин, — полученный именно против тирозинкиназы, успешно применяется для лечения содержащих рецептор HER2 опухолей молочных желез.
Кроме HER2 существует несколько десятков киназ, представляющих интерес в качестве специфических таргетных препаратов. Наиболее универсален среди них белок EGFR1, или HER1. Он активен практически во всех опухолях эпителиального происхождения. Сегодня в распоряжении клинических онкологов имеется два антитела и три низкомолекулярных антагониста EGFR. Их уже используют в рутинной практике лечения опухолей легкого, головы и шеи, толстой кишки и поджелудочной железы. Еще один пример — препарат гливек, направленный против тирозинкиназы ABL, активность которой лежит в основе хронического миелолейкоза и других онкологических заболеваний крови. При лечении некоторых разновидностей рака молочных желез используются ингибиторы серин-треониновой киназы mTOR.
Следующая группа мишеней — это тканеспецифические гормоны и их рецепторы. Среди таких препаратов наиболее известен тамоксифен. Его действие основано на связывании с рецептором эстрогенов при опухолях молочных желез. В лечении опухолей простаты широко применяют также ингибиторы ароматазы, осуществляющей конверсию андрогенов в эстрогены. Среди ангиогенных препаратов (напомним, что способность вызывать образование сосудов, т. е. ангиогенез, — характерная особенность опухоли) наибольшую известность получил препарат авастин — моноклональные антитела, способные связывать фактор роста эндотелия (VEGF).
За последние несколько лет найдены новые мишени таргетной терапии. Создание для них новых ингибиторов стремительно развивается, и в ближайшие годы можно ожидать выход в клинику новых эффективных таргетных препаратов.
Еще одно очень важное направление в лечении рака, основанное на знаниях молекулярно-генетических особенностей опухоли, — персонализированный подбор терапии. Иными словами, перед назначением того или иного препарата необходимо тщательное молекулярно-генетическое исследование, доказывающее наличие измененного гена — мишени для данного лекарства. Только в этом случае терапия может быть эффективной. И этот подход уже достаточно широко применяется в большинстве промышленно-развитых стран. Такие довольно трудоемкие работы открыли новые возможности для лечения опухолей молочных желез, легких, толстой кишки, желудка и других новообразований [15].
Анализ микроРНК в крови раковых больных позволяет по-другому проводить диагностику. Во-первых, спектр микроРНК в крови сходен с таковым в опухолях и отличен от нормы; во-вторых, этот метод неинвазивный, и забор материала для анализа не представляет никаких трудностей и может быть использован при самых ранних признаках появления болезни.
Поскольку микроРНК регулируют экспрессию ключевых генов, задействованных в канцерогенезе, эти некодирующие молекулы можно считать не только перспективными маркерами ранней диагностики и прогноза заболевания, но и мишенями терапевтического действия. Такими исследованиями охвачены самые распространенные формы рака — простаты, молочных желез, легкого и кишечника. Например, в опухоли простаты обнаружена одна микроРНК, активность которой более чем в 2000 раз превышает экспрессию в нормальных клетках; одновременный анализ четырех других видов микроРНК позволяет отличать опухоли простаты от доброкачественных образований и выявлять их достаточно высокую специфичность (до 90%).
В опухолях молочных желез и в крови больных имеется другая высокоактивная микроРНК и еще один вид микроРНК — в метастазах. Следует отметить, что эти опухоли бывают зависимыми и независимыми от эстрогенов и притом отличаются видами микроРНК. Следовательно, оценка уровня определенных микроРНК становится новым направлением в диагностике рака молочных желез.
При немелкоклеточном раке легкого в крови больных выявлено несколько видов микроРНК, характерных для самых ранних стадий болезни, а идентификация 11 других видов микроРНК может служить критерием продолжительности жизни больных. В крови больных колоректальным раком выявлено несколько видов микроРНК, позволяющих отличать его от рака желудка и кишечника, т. е. спектр активирующих микроРНК можно использовать как диагностический тест для идентификации опухолей кишечника. В последнее время стремительно растет объем информации о практической важности показателей экспрессии микроРНК в прогнозировании опухолей, в том числе и для больных с нейробластомой.
В ближайшем будущем можно ожидать создания наборов miR-микрочипов для диагностики некоторых видов опухолей.
***
Таким образом, молекулярная онкология как наука, возникшая в течение последних 50 лет, позволила не только вплотную подойти к пониманию молекулярных механизмов возникновения опухолей, но и привела к разработке новых и эффективных подходов в диагностике и терапии опухолей. Какие факты свидетельствуют об этом?
Во-первых, получены твердые доказательства того, что опухолевая клетка изменена генетически — это вновь возникшие мутации или мутации, индуцированные вирусным генетическим материалом за счет включения его в клеточный геном. Во-вторых, выяснилось, что активация онкогенов, инактивция генов-супрессоров и других генов, ассоциированных с канцерогенезом, могут возникать как за счет мутаций, так и за счет эпигенетических изменений. В-третьих, эпигенетические изменения, играющие ключевую роль в возникновении прогрессии опухолей, могут быть использованы в качестве биомаркеров для ранней диагностики опухолей, прогноза и мониторинга заболевания, предсказания устойчивости или чувствительности опухоли к химеотерапии, для выявления генов-мишеней лекарственной терапии.
Полностью расшифрован геном опухолей у больных раком различных локализаций, включая рак легких, рак молочной железы, колоректальный рак, а также некоторых неоплазий крови. Установлено, что 3–4% генома обладают кодирующим потенциалом и направляют синтез всех структурных белков клетки. Остальная часть генома выполняет регуляторные функции — синтез различных типов некодирующих РНК. Ключевая роль в регуляторных функциях принадлежит так называемым микроРНК, которые могут выполнять различные функции в канцерогенезе — активировать гены, или, наоборот, вызывать подавление их функций, они могут быть индикаторами стадийности опухолевого процесса, или фактором прогноза.
Главным достижением молекулярной онкологии последнего десятилетия оказалась разработка принципиального нового направления в терапии опухолей — так называемой таргетной терапии, которая направлена на подавление работы генов, активирующихся в опухолях. Такая терапия должна быть персонализирована — для каждого больного необходима выработка соответствующей схемы лечения. Исследования в этом направлении ведутся широким фронтом и за последние годы созданы десятки новых лекарств, которые существенно увеличивают продолжительность жизни больных раком.
Литература
1. Татосян А. Г. Онкогены // Канцерогенез / Ред. Д. Г. Заридзе, М., 2004. С. 103–124.
2. Зильбер Л. А., Ирлин И. С., Киселев Ф. Л. Эволюция вирусо-генетической теории возникновения опухолей. М., 1975.
3. Киселев Ф. Л., Павлиш О. А., Татосян А. Г. Молекулярные основы канцерогенеза у человека. М., 1990.
4. Копнин Б. П. Опухолевые супрессоры и мутаторные гены // Канцерогенез / Ред. Д. Г. Заридзе. М., 2004. С. 125–166.
5. Knudson A. G. Mutation and cancer: statistical study of retinoblastoma // Proc. Nat. Acad. Sci. USA. 1971. V. 68. P. 820–823.
6. Альтштейн А. Д. Вирусный канцерогенез и роль вирусов в возникновении опухолей у человека // Канцерогенез / Ред. Д. Г. Заридзе. М., 2004. С. 251–327.
7. Zur Hausen H. Infections causing human cancers. Weinheim; N. Y., 2011.
8. Hanahan D., Weinberg R. Hallmarks of cancer: next generation // Cell. 2011. V. 144. P. 646–674.
9. Garraway J., Linder E. Lessons from cancer genome // Cell. 2013. V. 153. P. 17–37.
10. Baylin S., Jones P. Functions of DNA methylation: islands, start sites, gene bodies and beyond // Nat. Rev. Genet. 2012. V. 13. P. 484–492.
11. Cedar H., Bergman Y. Linking DNA methylation and histone modification: patterns and paradigm // Nat. Rev. Genet. 2009. V. 10. P. 95–104.
12. Kouzarides T. Chromatin modifications and their functions // Cell. 2007. V. 128. P. 691–705.
13. Calin G., Croce C. MicroRNA signatures in human cancer // Nat. Rev Сancer. 2006. V. 6. P. 857–866.
14. Киселев Ф. Л., Имянитов Е. Н., Киселева Н. П., Левина Е. С. Молекулярная онкология: от вирусной теории к лечению рака. М., 2013.
15. Imjanitov E., Moiseyеnko V. Drug therapy for hereditary cancers // Нered. Cancer. Clin. Pract. 2011. V. 9. P. 5–7.
1 О том, как в тюремных застенках эта идея пришла в голову Л. А. Зильберу, можно прочитать в книге: Киселев Л. Л., Левина Е. С. Лев Александрович Зильбер. М., 2004.
2 Лауреаты Нобелевской премии 1989 года. По физиологии или медицине — М. Бишоп и Х. Вармус. Киселев Л. Л. // Природа. 1990. № 1. С. 96–98 (DjVu, 4,6 Мб).
3 Вирус саркомы Меркеля был выделен позднее, чем остальные, в 2008 г.
4 Подробнее см.: Киселев Ф. Л., Боринская С. А. Вакцина против рака // Природа. 2007. № 3. С. 52–58 (PDF, 4 Мб).
-
Неплохо было бы особый акцент сделать на внутренней молекулярной кухне. При репликации ф. Оказакки недореплицированы основным репл. комплексом. Их потом достраивает репарационная полимераза, склонная к ошибкам. Поэтому рак, в основном, -- болезнь пожилых: больше делений -- больше ошибок. А также: апуринизация, тиминовые димеры, аддукты, интеркаляты, метил- тимин, NO6- аденин из-за атаки NO и др. RO'. Таутомеры; и самое экзотическое -- спонтанная диссоциация Н2О возле ДНК. Я это перечислил к тому, что слишком много случайных факторов, которые (несовершенства) вшиты в саму репликативную машинерию. А если взять клональные войны в самой опухоли, то сложность взаимодействий возрастает.












Функции гена-супрессора Rb в клетках. CDK4/6 — одна из циклин-зависимых киназ, регулирующих активность этого гена; p16 — ингибитор киназы; E2F — транскрипционный фактор, играющий ключевую роль в транскрипции. Внизу приведены основные фазы клеточного цикла: G0 — клеточного покоя, G1 — подготовки к синтезу ДНК, S — репликации ДНК, G2 — активного роста клеток, М — фаза деления клетки (митоз)